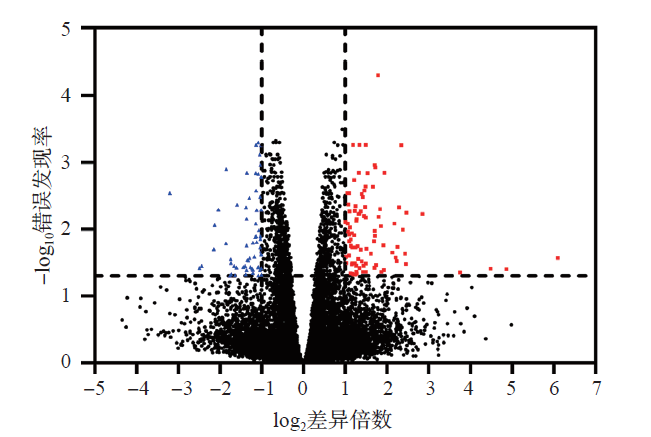
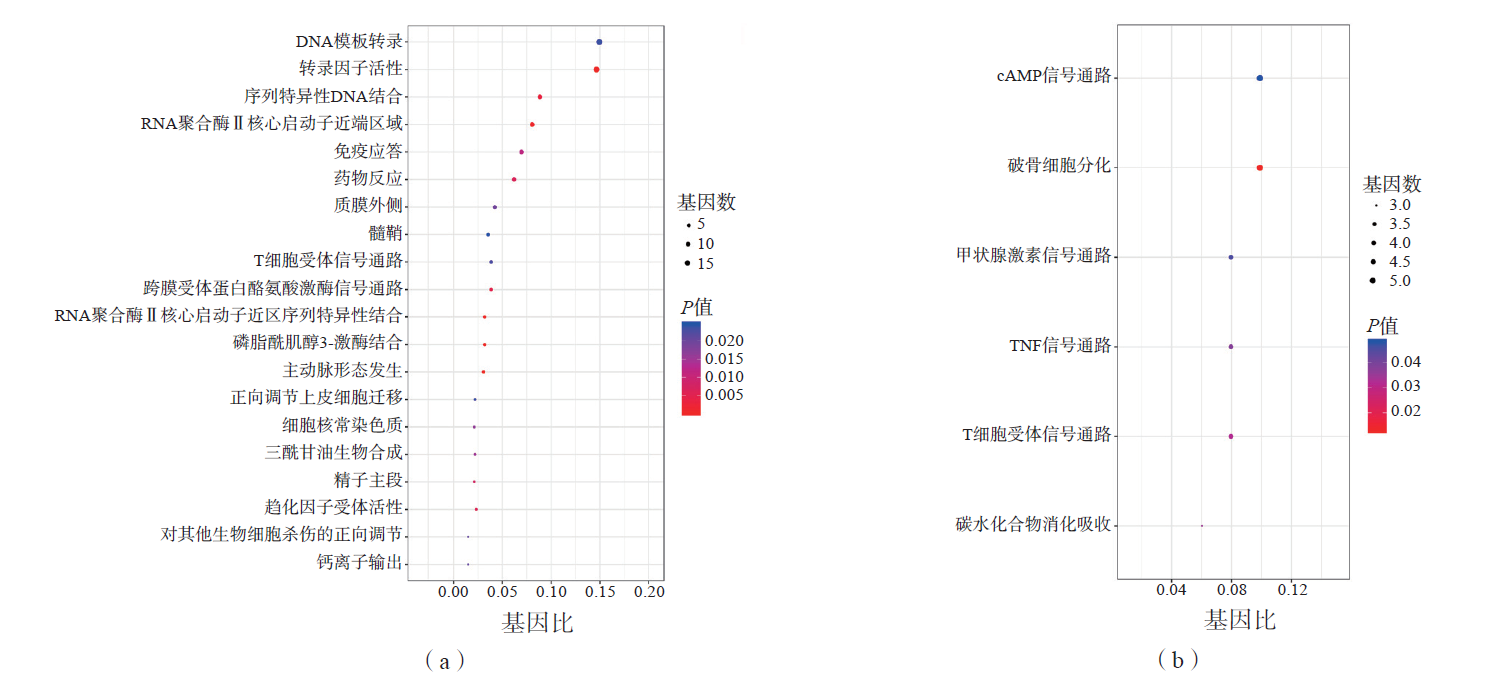
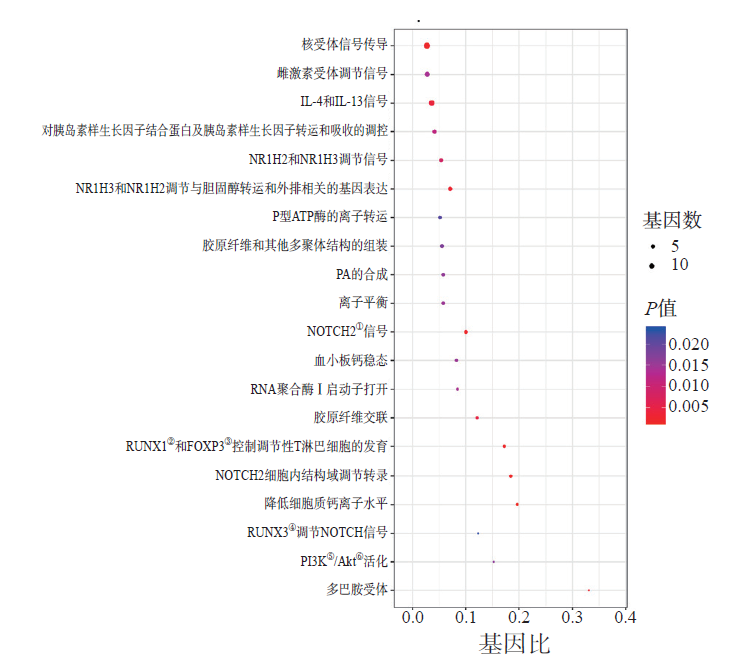
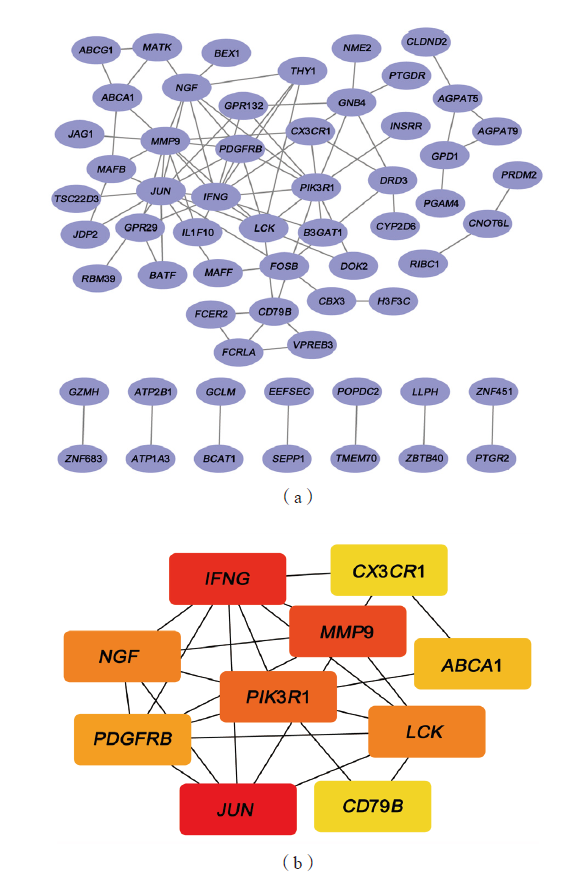
Laboratory Medicine ›› 2023, Vol. 38 ›› Issue (1): 32-38.DOI: 10.3969/j.issn.1673-8640.2023.01.007
Previous Articles Next Articles
CHEN Xuewei, LI Yirong( )
)
Received:2021-08-31
Revised:2022-11-19
Online:2023-01-30
Published:2023-03-15
CLC Number:
CHEN Xuewei, LI Yirong. Candidate biomarkers in metabolic syndrome based on GEO database[J]. Laboratory Medicine, 2023, 38(1): 32-38.
Add to citation manager EndNote|Ris|BibTeX
URL: https://www.shjyyx.com/EN/10.3969/j.issn.1673-8640.2023.01.007
| [1] |
BORGHI C, AGABITI-ROSEI E, JOHNSON R J, et al. Hyperuricaemia and gout in cardiovascular,metabolic and kidney disease[J]. Eur J Intern Med, 2020, 80:1-11.
DOI URL |
| [2] |
WANG C H, LUNDH M, FU A, et al. CRISPR-engineered human brown-like adipocytes prevent diet-induced obesity and ameliorate metabolic syndrome in mice[J]. Sci Transl Med, 2020, 12(558):eaaz8664.
DOI URL |
| [3] |
MASSILLO C, DUCA R B, LACUNZA E, et al. Adipose tissue from metabolic syndrome mice induces an aberrant miRNA signature highly relevant in prostate cancer development[J]. Mol Oncol, 2020, 14(11):2868-2883.
DOI URL |
| [4] | HU C, JIA W. Multi-omics profiling:the way towards precision medicine in metabolic diseases[J]. J Mol Cell Biol, 2021. [Epub ahead of print] |
| [5] | 刘晓妮, 王颖, 李桃桃, 等. 2型糖尿病合并代谢综合征患者胰岛素抵抗指数与慢性炎症指标相关性研究[J]. 检验医学, 2019, 34(9):826-830. |
| [6] |
ULVEN S M, HOLVEN K B, RUNDBLAD A, et al. An isocaloric nordic diet modulates RELA and TNFRSF1A gene expression in peripheral blood mononuclear cells in individuals with metabolic syndrome-a SYSDIET sub-study[J]. Nutrients, 2019, 11(12):2932.
DOI URL |
| [7] |
FRANCESCHI C, GARAGNANI P, PARINI P, et al. Inflammaging:a new immune-metabolic viewpoint for age-related diseases[J]. Nat Rev Endocrinol, 2018, 14(10):576-590.
DOI |
| [8] |
CAI J, XU M, ZHANG X, et al. Innate immune signaling in nonalcoholic fatty liver disease and cardiovascular diseases[J]. Annu Rev Pathol, 2019, 14:153-184.
DOI PMID |
| [9] |
DARYABOR G, ATASHZAR M R, KABELITZ D, et al. The effects of type 2 diabetes mellitus on organ metabolism and the immune system[J]. Front Immunol, 2020, 11:1582.
DOI PMID |
| [10] |
KEWCHAROENWONG C, SAENWONGSA W, WILLCOCKS S J, et al. Glibenclamide alters interleukin-8 and interleukin-1β of primary human monocytes from diabetes patients against Mycobacterium tuberculosis infection[J]. Tuberculosis (Edinb), 2020, 123:101939.
DOI URL |
| [11] | SCHNEIDER-MATYKA D, SZKUP M, OWCZAREK A J, et al. The relationship between the IFNG (rs2430561) polymorphism and metabolic syndrome in perimenopausal women[J]. Medicina (Kaunas), 2020, 56(8):384. |
| [12] |
JAISWAL N, AGRAWAL S, AGRAWAL A. High fructose-induced metabolic changes enhance inflammation in human dendritic cells[J]. Clin Exp Immunol, 2019, 197(2):237-249.
DOI PMID |
| [13] |
LIU J, GUO Z, ZHANG Y, et al. LCK inhibitor attenuates atherosclerosis in ApoE-/- mice via regulating T cell differentiation and reverse cholesterol transport[J]. J Mol Cell Cardiol, 2020, 139:87-97.
DOI URL |
| [14] |
LIN W, LIU H, TANG Y, et al. The development and controversy of competitive endogenous RNA hypothesis in non-coding genes[J]. Mol Cell Biochem, 2021, 476(1):109-123.
DOI |
| [15] |
DOLATI S, SHAKOURI S K, DOLATKHAH N, et al. The role of exosomal non-coding RNAs in aging-related diseases[J]. Biofactors, 2021, 47(3):292-310.
DOI PMID |
| [16] |
ORTIZ-DOSAL A, RODIL-GARCÍA P, SALAZAR-OLIVO L A. Circulating microRNAs in human obesity:a systematic review[J]. Biomarkers, 2019, 24(6):499-509.
DOI URL |
| [17] | HUANG Y, ZHAO L, YAN Y, et al. PBMCs to stress-associated miR-18a-5p and miR-22-3p ratios as new indicators of metabolic syndrome[J]. Biomed Res Int, 2020, 2020:8159342. |
| [18] |
BROZZI F, REGAZZI R. Circular RNAs as novel regulators of β-cell functions under physiological and pathological conditions[J]. Int J Mol Sci, 2021, 22(4):1503.
DOI URL |
| [19] |
ZAIOU M. Circular RNAs as potential biomarkers and therapeutic targets for metabolic diseases[J]. Adv Exp Med Biol, 2019, 1134:177-191.
DOI PMID |
| [1] | LI Liangshan, ZHAO Hu, WANG Shiwen. Screening potential prognostic biomarkers of colorectal cancer based on weighted gene co-expression network analysis [J]. Laboratory Medicine, 2023, 38(9): 825-832. |
| [2] | SUN Zepeng, WANG Hongbin, WANG Jiandong, SONG Dewei, XIAO Peng. Analysis and progress of peptide and protein biomarker methodology for myocardial injury [J]. Laboratory Medicine, 2023, 38(8): 784-789. |
| [3] | WANG Tengfei, CHEN Shan, AN Hebing, ZHOU Lei. Expressions and roles of lncRNA CRNDE and miR-384 in peripheral blood of patients with acute leukemia [J]. Laboratory Medicine, 2023, 38(7): 686-691. |
| [4] | ZHANG Min, WANG Binyu, CHI Weiqun, LIU Yu. Research progress of exosomal non-coding RNA as biomarkers for disease diagnosis [J]. Laboratory Medicine, 2023, 38(6): 594-598. |
| [5] | GAO Feng. Clinical application of novel tumor biomarkers:prospects and challenges [J]. Laboratory Medicine, 2023, 38(4): 303-306. |
| [6] | CHEN Hui, CHEN Guanghui, LIANG Yingliang, WANG Wandang, YIN Zhijun. Screening hub genes and diagnostic analysis of gestational diabetes mellitus based on GEO database [J]. Laboratory Medicine, 2023, 38(3): 276-281. |
| [7] | ZHOU Furong, LI Yanzhu, LIU Yonggan. Application of lncRNA SNP in colorectal cancer susceptibility prediction and prognosis assessment [J]. Laboratory Medicine, 2023, 38(12): 1206-1210. |
| [8] | LU Qiyuan, LU Jianhua. Biomarker research progress for the diagnosis of periprosthetic joint infection [J]. Laboratory Medicine, 2023, 38(10): 997-1002. |
| [9] | WU Yating, LI Zhuolin, LEI Yan, JIA Ruxue, ZHANG Shenghang, WANG Shuiliang. Research progress of miRNA in urine as a biomarker for common malignant tumors [J]. Laboratory Medicine, 2023, 38(1): 94-99. |
| [10] | WANG Rui, LI Zhaoyan, ZHAO Aiguang. Application of circulating tumor DNA detection in the diagnosis and treatment of gastric cancer [J]. Laboratory Medicine, 2022, 37(9): 877-881. |
| [11] | WU Yanqian, YU Chong, SHEN Lu, ZHENG Haoran, HONG Yeting. Research progress of body fluid piRNA as a potential biomarker of disease [J]. Laboratory Medicine, 2022, 37(8): 782-786. |
| [12] | FENG Xueqing, JU Yi, LI Qing, JIN Zhonggan. Research progress of electrochemical aptasensors for the determination of biomarkers [J]. Laboratory Medicine, 2022, 37(5): 477-482. |
| [13] | CHEN Liang, LI Lin, LI Xia, ZHANG Yan, WU Jiongrui, GAO Yiming. Correlation between metabolic syndrome and moderate-to-severe periodontitis complicated with psoriasis [J]. Laboratory Medicine, 2022, 37(2): 126-129. |
| [14] | CHI Wenjing, DING Li, LIU Yixin, YANG Feng, LIU Tao, CONG Jianhua, GAO Zhenhui, ZHAO Hu, ZHANG Yanmei. Correlation analysis of Helicobacter pylori colonization level and metabolic syndrome in physical examination population in Shanghai [J]. Laboratory Medicine, 2022, 37(11): 1034-1038. |
| [15] | HUANG Xiaofeng, FAN Xueming, YAO Tianyue, YUAN Wenhua, ZHAO Zhiyun, SONG Yunxiao. Correlation between red cell distribution width and prostate cancer [J]. Laboratory Medicine, 2021, 36(6): 590-595. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||