
检验医学 ›› 2026, Vol. 41 ›› Issue (2): 133-143.DOI: 10.3969/j.issn.1673-8640.2026.02.006
王亚琼1, 应令雯2, 陈瑶2, 姚如恩3, 娄丹1, 卢亚亚1, 李娟2, 王秀敏2( )
)
收稿日期:2025-03-03
修回日期:2025-12-29
出版日期:2026-02-28
发布日期:2026-03-06
通讯作者:
王秀敏,E-mail:wangxiumin1019@126.com。
作者简介:王亚琼,女,1986年生,主治医师,主要从事儿童生长发育障碍性疾病的基础和临床研究。
基金资助:
WANG Yaqiong1, YING Lingwen2, CHEN Yao2, YAO Ruen3, LOU Dan1, LU Yaya1, LI Juan2, WANG Xiumin2()
Received:2025-03-03
Revised:2025-12-29
Online:2026-02-28
Published:2026-03-06
摘要:
目的 探讨转运蛋白复合体亚单位2(TRAPPC2)基因变异致X-连锁迟发性脊椎骨骺发育不良(SEDT)家系的临床表型和遗传学特点。方法 选取2019年1月—2024年9月上海交通大学医学院附属上海儿童医学中心确诊的3个X-连锁SEDT家系。收集家系成员的临床资料,并进行全外显子组测序(WES)和生物信息学分析,采用Sanger测序对可疑突变进行验证。参照美国医学遗传学与基因组学学会(ACMG)相关指南对变异进行评级。结果 3个家系的先证者均为男性,均因生长迟缓就诊,脊柱X线片示椎体扁平、前部上下缘凹陷、中后部呈驼峰样突起改变。家系1和家系2的先证者TRAPPC2基因(NM_001011658.4)均存在半合子变异c.271_275del(p.Gln91Argfs*9),为致病性变异;先证者母亲该位点均存在杂合突变,父亲未检测到该突变。家系3先证者TRAPPC2基因(NM_001011658.4)存在半合子变异c.191_192delTG(p.Val64Glyfs*24),其父母未发现该位点存在任何突变,考虑为假定新发变异;行重组人生长激素(rhGH)治疗,疗效欠佳。结论 TRAPPC2基因变异所致的SEDT呈晚发性、进行性特征,可致不成比例的身材矮小和关节过早退化。对于青少年时期起病的患儿,若合并上下部量异常或指尖距不匹配,需警惕SEDT。WES检测有助于明确病因。
中图分类号:
王亚琼, 应令雯, 陈瑶, 姚如恩, 娄丹, 卢亚亚, 李娟, 王秀敏. TRAPPC2基因变异致X-连锁迟发性脊椎骨骺发育不良的3个家系分析[J]. 检验医学, 2026, 41(2): 133-143.
WANG Yaqiong, YING Lingwen, CHEN Yao, YAO Ruen, LOU Dan, LU Yaya, LI Juan, WANG Xiumin. Analysis of 3 families with X-linked spondyloepiphyseal dysplasia tarda caused by TRAPPC2 gene variation[J]. Laboratory Medicine, 2026, 41(2): 133-143.
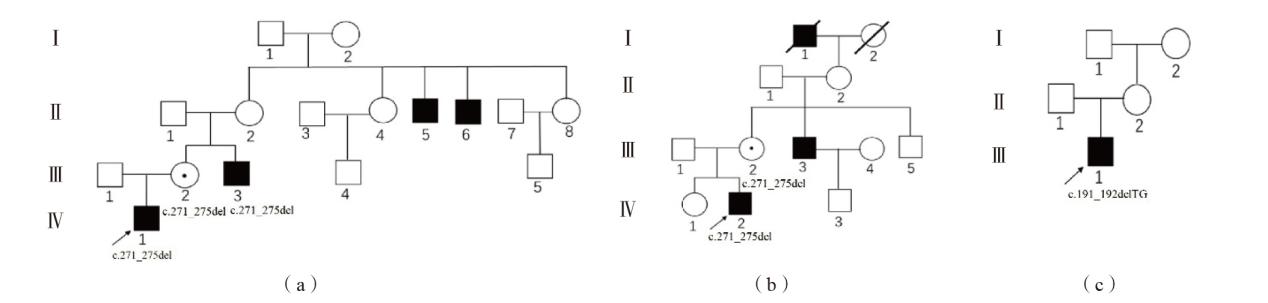
图1 3个家系的家系图 注:(a)家系1;(b)家系2;(c)家系3;Ⅰ~Ⅳ为家系代数;□正常男性、○正常女性、■男性患者、↗先证者、☉女性携带者、∕死亡个体。
| 基因名称 | 正向引物(5'~3') | 反向引物(5'~3') |
|---|---|---|
| TRAPPC2-E3 | GAATTCTACACTTCCCATTAGTC | TATCTGTCCAGATCTTCCAGTTC |
| TRAPPC2-E4 | TGGCTGTTTCTGTTGAGATGT | GAGCCTGGCATCTATGTTCC |
| TRAPPC2-E5 | TCCATCACAAATACACTGCATGT | GGTAACTACCATGAAGTTGTGTTCC |
| TRAPPC2-E6 | CAGAAACTTAAGATTTGTCAGC | TGACATGAGACAGAATGTACTA |
表1 引物序列
| 基因名称 | 正向引物(5'~3') | 反向引物(5'~3') |
|---|---|---|
| TRAPPC2-E3 | GAATTCTACACTTCCCATTAGTC | TATCTGTCCAGATCTTCCAGTTC |
| TRAPPC2-E4 | TGGCTGTTTCTGTTGAGATGT | GAGCCTGGCATCTATGTTCC |
| TRAPPC2-E5 | TCCATCACAAATACACTGCATGT | GGTAACTACCATGAAGTTGTGTTCC |
| TRAPPC2-E6 | CAGAAACTTAAGATTTGTCAGC | TGACATGAGACAGAATGTACTA |
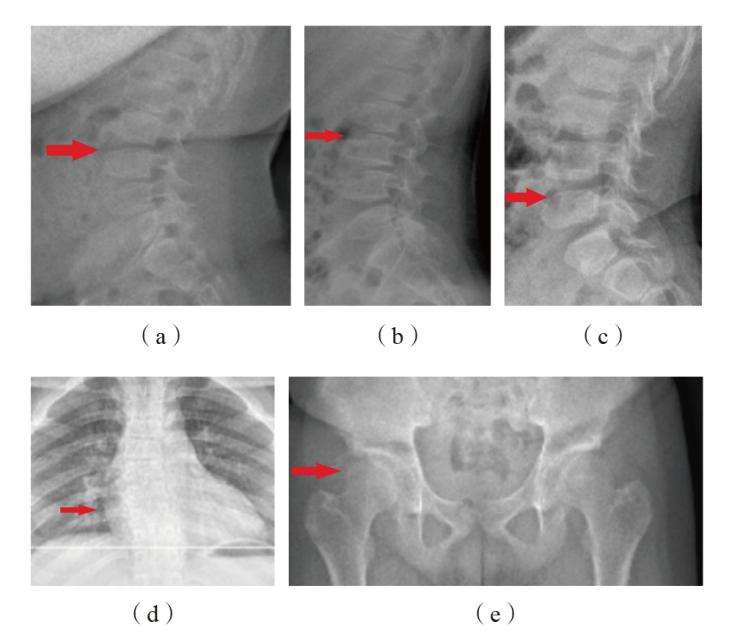
图2 3例SEDT患儿X线检查结果 注:(a)(b)(c)分别为家系1、家系2、家系3中SEDT患儿(先证者)的脊柱侧位X线片,均表现为胸腰椎椎体前部上下缘凹陷、中后部呈驼峰样突起改变;(d)家系2中SEDT患儿(先证者)的脊柱正位X线片,提示胸椎侧弯;(e)家系2中SEDT患儿(先证者)的骨盆正位X线片,提示双侧髋臼缘毛糙、双侧股骨头内侧偏扁。
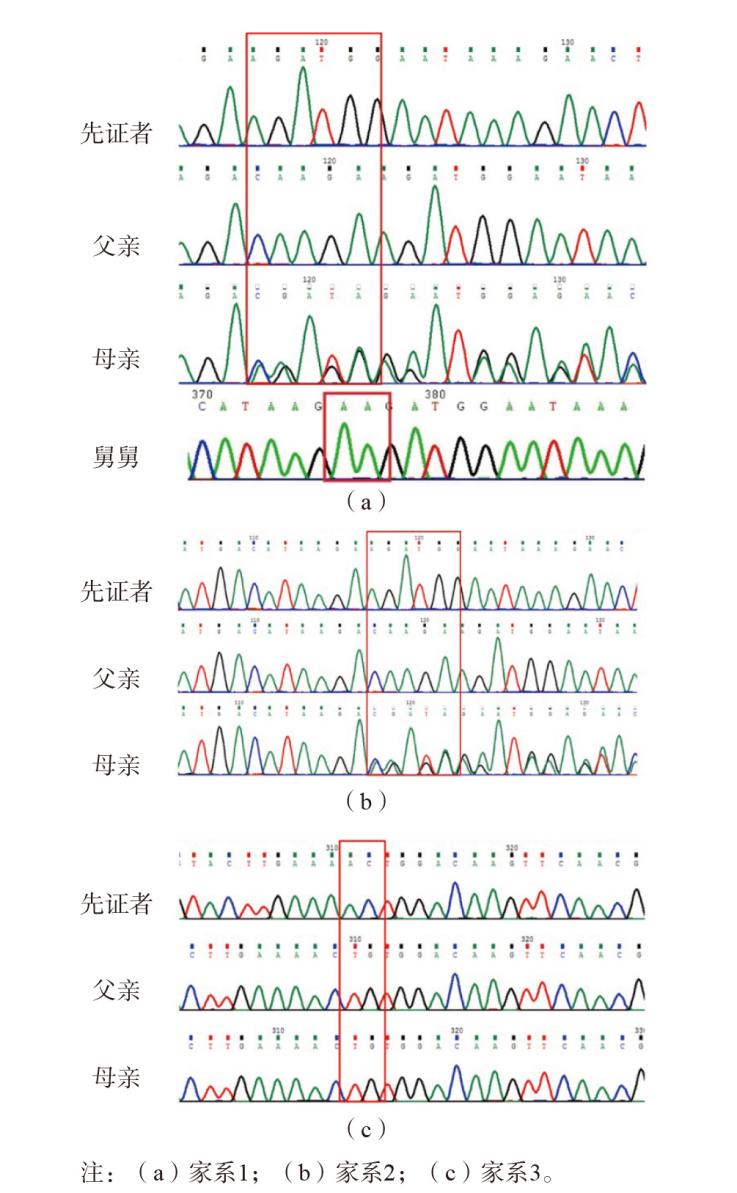
图3 3个家系Sanger测序图 注:(a)家系1;(b)家系2;(c)家系3。
| 位置 | 核苷酸变化 | 变异类型 | 年龄/岁 | 身高/cm | 指间距/cm | 骨关节症状 | 文献 |
|---|---|---|---|---|---|---|---|
| 内含子2 | c.-19-2A>G | 剪接突变 | GEDEON等[ | ||||
| 内含子2 | c.-19-2A > G. | 剪接突变 | 6 | 109(-1.91s) | 无 | LI等[ | |
| 内含子2 | c.-19-2A>C | 剪接突变 | 19 | 140(-5.36s) | 159 | ①+② | WANG等[ |
| 内含子2~ 外显子3 | 1763 bp片段缺失 | 缺失突变 | 23 | 154(-3.07s) | 165 | ① | MATSUI等[ |
| 外显子3 | c.1A>T | 错义突变 | 49 | 138(-5.69s) | 152 | ③ | WON等[ |
| 外显子3 | c.6delT | 缺失突变 | GOMES等[ | ||||
| 外显子3 | c.40delG | 缺失突变 | 15 | 147(-3.54s) | ① | WON等[ | |
| 外显子3 | c.53_54delTT | 缺失突变 | 成人 | 160(-2.08s) | ③ | GEDEON等[ | |
| 外显子3 | c.61G>T | 无义突变 | 30 | 147(-4.21s) | 165 | ①+③ | XIA等[ |
| 外显子3 | 外显子3缺失 | 缺失突变 | 16 | 139(-5.21s) | 160 | ①+②+③ | GEDEON等[ |
| 外显子3 | c.91A>T | 无义突变 | 成人 | 135(-6.18s) | ①+③ | LOU等[ | |
| 外显子3 | c.91A>T | 无义突变 | 28 | 148(-4.05s) | 152 | ①+③ | 张闻宇等[ |
| 内含子3 | c.93+1G>A | 剪接突变 | 9岁 | 112(-4.03s) | 无 | ADACHI等[ | |
| 内含子3 | c.93+5G>A | 剪接突变 | 成人 | 156(-2.74s) | 176 | ③ | GEDEON等[ |
| 内含子3 | c.93+5G>A | 剪接突变 | 14 | 148(-2.51s) | 无 | RYU等[ | |
| 内含子3 | c.93+5G>A | 剪接突变 | 11 | 134(-1.71s) | ①+③ | WU等[ | |
| 内含子3 | c.93+5G>C | 剪接突变 | 成人 | 140(-5.36s) | 165 | 无 | WANG等[ |
| 内含子3 | c.94-2A>G | 剪接突变 | 13 | 128(-4.09s) | 134 | ③ | FUKUMA等[ |
| 外显子4-6 | 外显子4~6缺失 | 缺失突变 | FIEDLER等[ | ||||
| 外显子4-6 | 2.5 kb片段缺失 | 缺失突变 | 24 | 155(-2.90s) | ① | 刘宇等[ | |
| 外显子4~ 内含子5 | c.197_324+121del693bp | 缺失突变 | 12 | 115(-4.90s) | 122 | 无 | TAKAGI等[ |
| 外显子4 | c.94delG | 缺失突变 | 27 | 140(-5.36s) | 151 | ①+③ | 黄颖等[ |
| 外显子4 | c.100delC | 缺失突变 | FIEDLER等[ | ||||
| 外显子4 | c.115delC | 缺失突变 | 9 | 115(-3.52s) | 109 | ② | 王莉莉等[ |
| 外显子4 | c.137_138del | 缺失突变 | 23 | 151(-3.56s) | ② | YUNIS等[ | |
| 外显子 4 | c.139G>T | 错义突变 | GEDEON等[ | ||||
| 外显子 4 | c.157-158delAT | 缺失突变 | 成人 | 140(-5.36s) | 155 | ② | GEDEON等[ |
| 外显子4 | c.157-158delAT | 缺失突变 | 22 | 140(-5.36s) | 155 | ②+③ | GEDEON等[ |
| 外显子4 | c.167C>A | 无义突变 | 47 | 160(-2.08s) | 163~174 | ③ | FIEDLER等[ |
| 外显子 4 | c.182T>A | 无义突变 | GEDEON等[ | ||||
| 外显子4 | c.183_184delGA | 缺失突变 | FIEDLER等[ | ||||
| 外显子 4 | c.191-192delTG | 缺失突变 | 成人 | 153(-3.23s) | 164 | ①+③ | GEDEON等[ |
| 外显子 4 | c.191-192delTG | 缺失突变 | 9 | 123 (-2.42s) | 130 | 无 | 本研究家系3 |
| 外显子4 | c.209G>A | 无义突变 | 45 | 149(-3.89s) | ①+② | 朱海燕等[ | |
| 外显子 4 | c.209G>A | 无义突变 | ①+③ | CAO等[ | |||
| 外显子4 | c.209G>A | 无义突变 | 31 | 148(-4.05s) | 164 | ③ | TAMHANKAR等[ |
| 外显子4 | c.210G>A | 无义突变 | CHRISTIE等[ | ||||
| 外显子4 | c.216_217del | 缺失突变 | 26 | 137(-5.80s) | 144 | ① | ZHANG等[ |
| 外显子4 | c.218C>T | 错义突变 | 成人 | 137(-5.85s) | ③ | 王莉等[ | |
| 外显子4 | c.218C>T | 错义突变 | 成人 | 146(-4.38s) | 158 | ① | ZHOU等[ |
| 外显子 4 | c.218C>T | 错义突变 | GEDEON等[ | ||||
| 内含子 4 | c.238+1A>G | 剪接突变 | 38 | 135(-6.18s) | 155 | ① | GUO等[ |
| 内含子4 | c.238+4T>C | 剪接突变 | 14 | 127(-4.27s) | 130 | ①+② | SHAW等[ |
| 外显子4 | C.239A>G | 错义突变 | 37 | 159(-2.25s) | ①+③ | LIN等[ | |
| 内含子4 | c.239-10_239-7delATTA | 剪接突变 | GEDEON等[ | ||||
| 内含子4 | c.239-11_239-4delAATTATTT | 剪接突变 | GEDEON等[ | ||||
| 内含子4 | c.239-11_239-9delAAT | 剪接突变 | 42 | 148(-4.03s) | ①+③ | DAVIS等[ | |
| 外显子5 | c.241_242delAT | 缺失突变 | GEDEON等[ | ||||
| 外显子5 | c.241_242delAT | 缺失突变 | 15 | ② | MUMM等[ | ||
| 外显子5 | c.248T>C | 错义突变 | GRUNEBAUM等[ | ||||
| 外显子5 | c.260A>C | 错义突变 | 13 | 126(-1.89s) | 138 | 无 | ZHANG等[ |
| 外显子 5 | c.262_266delGACAT | 缺失突变 | GEDEON等[ | ||||
| 外显子5 | c.262_266delGACAT | 缺失突变 | 25 | 143(-4.87s) | 154 | ③ | 孔祥东等[ |
| 外显子5 | c.267_271delAAGAC | 缺失突变 | 26 | 158(-2.41s) | 178 | ③ | FIEDLER等[ |
| 外显子5 | c.267_27ldelAAGAC | 缺失突变 | 10 | 120(-3.21s) | 无 | 李娟等[ | |
| 外显子 5 | c.271 C>T | 无义突变 | GEDEON等[ | ||||
| 外显子5 | c.271C>T | 无义突变 | 13 | 139(-2.73s) | 130 | 无 | 孟婕等[ |
| 外显子 5 | c.271-275delCAAGA | 缺失突变 | GEDEON等[ | ||||
| 外显子5 | c.271-275delCAAGA | 缺失突变 | 29 | 135 (-6.18s) | 无 | 黄欢等[ | |
| 外显子5 | C.271-275delCAAGA | 缺失突变 | 37 | 118(-8.97s) | ①+③ | 徐贵江等[ | |
| 外显子5 | C.271-275del | 缺失突变 | 11 | 141(-1.11s) | 147 | 无 | 本研究家系1 |
| 外显子5 | C.271-275del | 缺失突变 | 11 | 138(-1.14s) | 145 | ② | 本研究家系2 |
| 外显子 5 | c.272-273delAA | 缺失突变 | GEDEON等[ | ||||
| 外显子5 | c.293delT | 缺失突变 | 27 | 112(-9.95s) | 142 | ①+③ | XIAO等[ |
| 外显子5 | c.320dupT | 插入突变 | 16 | 147(-4.02s) | 164 | GEDEON等[ | |
| 内含子5 | c.325-2A>C | 剪接突变 | 成人 | 152(-3.39s) | 168 | ① | GEDEON等[ |
| 内含子5 | c.325-10_3254delTCTTTCCinsAA | 剪接突变 | GEDEON等[ | ||||
| 内含子5~ 外显子 6 | 1 330 bp片段缺失 | 缺失突变 | GEDEON等[ | ||||
| 内含子5~ 外显子 6 | 1 371~1 445 bp片段缺失 | 缺失突变 | CHRISTIE等[ | ||||
| 内含子5~ 外显子 6 | 750 bp片段缺失 | 缺失突变 | CHRISTIE等[ | ||||
| 内含子5~ 外显子 6 | c.325-2_335delAGTTTTCAATGAA | 缺失突变 | 16 | 143(-4.61s) | 151 | 无 | 麻宏伟等[ |
| 内含子5~ 外显子6 | 1.335 kb片段缺失 | 缺失突变 | 20 | 157(-2.57s) | 172 | ① | SHAW等[ |
| 外显子6 | 外显子6缺失 | 缺失突变 | 成人 | 153(-3.31s) | 171 | ③ | GEDEON等[ |
| 外显子6 | c.328delT | 缺失突变 | FIEDLER等[ | ||||
| 外显子6 | c.329C>A | 无义突变 | 16 | 143(-4.61s) | 153 | 无 | SHI等[ |
| 外显子6 | c.333_336delGAAT | 缺失突变 | 14 | 142(-3.32s) | ② | SHAW等[ | |
| 外显子6 | c.345_346delTG | 缺失突变 | FIEDLER等[ | ||||
| 外显子6 | c.370-371insA | 插入突变 | 20 | 153(-3.23s) | 167 | 无 | XIA等[ |
| 外显子6 | c.387delA | 缺失突变 | 16 | 147(-3.97s) | ① | BAR-YOSEF等[ | |
| 外显子6 | c. 389 T>A | 错义突变 | GEDEON等[ | ||||
| 外显子6 | c.391C>T | 插入突变 | 14 | 146(-2.76s) | 170 | 无 | TAKAHASHI等[ |
| 外显子6 | c.391_392del | 缺失突变 | 28 | ①+② | HE等[ | ||
| 外显子6 | c.406A>T | 错义突变 | 15 | 134(-5.44s) | 144 | ①+② | YASAR等[ |
表2 SEDT患者TRAPPC2基因变异位置、类型和临床表型
| 位置 | 核苷酸变化 | 变异类型 | 年龄/岁 | 身高/cm | 指间距/cm | 骨关节症状 | 文献 |
|---|---|---|---|---|---|---|---|
| 内含子2 | c.-19-2A>G | 剪接突变 | GEDEON等[ | ||||
| 内含子2 | c.-19-2A > G. | 剪接突变 | 6 | 109(-1.91s) | 无 | LI等[ | |
| 内含子2 | c.-19-2A>C | 剪接突变 | 19 | 140(-5.36s) | 159 | ①+② | WANG等[ |
| 内含子2~ 外显子3 | 1763 bp片段缺失 | 缺失突变 | 23 | 154(-3.07s) | 165 | ① | MATSUI等[ |
| 外显子3 | c.1A>T | 错义突变 | 49 | 138(-5.69s) | 152 | ③ | WON等[ |
| 外显子3 | c.6delT | 缺失突变 | GOMES等[ | ||||
| 外显子3 | c.40delG | 缺失突变 | 15 | 147(-3.54s) | ① | WON等[ | |
| 外显子3 | c.53_54delTT | 缺失突变 | 成人 | 160(-2.08s) | ③ | GEDEON等[ | |
| 外显子3 | c.61G>T | 无义突变 | 30 | 147(-4.21s) | 165 | ①+③ | XIA等[ |
| 外显子3 | 外显子3缺失 | 缺失突变 | 16 | 139(-5.21s) | 160 | ①+②+③ | GEDEON等[ |
| 外显子3 | c.91A>T | 无义突变 | 成人 | 135(-6.18s) | ①+③ | LOU等[ | |
| 外显子3 | c.91A>T | 无义突变 | 28 | 148(-4.05s) | 152 | ①+③ | 张闻宇等[ |
| 内含子3 | c.93+1G>A | 剪接突变 | 9岁 | 112(-4.03s) | 无 | ADACHI等[ | |
| 内含子3 | c.93+5G>A | 剪接突变 | 成人 | 156(-2.74s) | 176 | ③ | GEDEON等[ |
| 内含子3 | c.93+5G>A | 剪接突变 | 14 | 148(-2.51s) | 无 | RYU等[ | |
| 内含子3 | c.93+5G>A | 剪接突变 | 11 | 134(-1.71s) | ①+③ | WU等[ | |
| 内含子3 | c.93+5G>C | 剪接突变 | 成人 | 140(-5.36s) | 165 | 无 | WANG等[ |
| 内含子3 | c.94-2A>G | 剪接突变 | 13 | 128(-4.09s) | 134 | ③ | FUKUMA等[ |
| 外显子4-6 | 外显子4~6缺失 | 缺失突变 | FIEDLER等[ | ||||
| 外显子4-6 | 2.5 kb片段缺失 | 缺失突变 | 24 | 155(-2.90s) | ① | 刘宇等[ | |
| 外显子4~ 内含子5 | c.197_324+121del693bp | 缺失突变 | 12 | 115(-4.90s) | 122 | 无 | TAKAGI等[ |
| 外显子4 | c.94delG | 缺失突变 | 27 | 140(-5.36s) | 151 | ①+③ | 黄颖等[ |
| 外显子4 | c.100delC | 缺失突变 | FIEDLER等[ | ||||
| 外显子4 | c.115delC | 缺失突变 | 9 | 115(-3.52s) | 109 | ② | 王莉莉等[ |
| 外显子4 | c.137_138del | 缺失突变 | 23 | 151(-3.56s) | ② | YUNIS等[ | |
| 外显子 4 | c.139G>T | 错义突变 | GEDEON等[ | ||||
| 外显子 4 | c.157-158delAT | 缺失突变 | 成人 | 140(-5.36s) | 155 | ② | GEDEON等[ |
| 外显子4 | c.157-158delAT | 缺失突变 | 22 | 140(-5.36s) | 155 | ②+③ | GEDEON等[ |
| 外显子4 | c.167C>A | 无义突变 | 47 | 160(-2.08s) | 163~174 | ③ | FIEDLER等[ |
| 外显子 4 | c.182T>A | 无义突变 | GEDEON等[ | ||||
| 外显子4 | c.183_184delGA | 缺失突变 | FIEDLER等[ | ||||
| 外显子 4 | c.191-192delTG | 缺失突变 | 成人 | 153(-3.23s) | 164 | ①+③ | GEDEON等[ |
| 外显子 4 | c.191-192delTG | 缺失突变 | 9 | 123 (-2.42s) | 130 | 无 | 本研究家系3 |
| 外显子4 | c.209G>A | 无义突变 | 45 | 149(-3.89s) | ①+② | 朱海燕等[ | |
| 外显子 4 | c.209G>A | 无义突变 | ①+③ | CAO等[ | |||
| 外显子4 | c.209G>A | 无义突变 | 31 | 148(-4.05s) | 164 | ③ | TAMHANKAR等[ |
| 外显子4 | c.210G>A | 无义突变 | CHRISTIE等[ | ||||
| 外显子4 | c.216_217del | 缺失突变 | 26 | 137(-5.80s) | 144 | ① | ZHANG等[ |
| 外显子4 | c.218C>T | 错义突变 | 成人 | 137(-5.85s) | ③ | 王莉等[ | |
| 外显子4 | c.218C>T | 错义突变 | 成人 | 146(-4.38s) | 158 | ① | ZHOU等[ |
| 外显子 4 | c.218C>T | 错义突变 | GEDEON等[ | ||||
| 内含子 4 | c.238+1A>G | 剪接突变 | 38 | 135(-6.18s) | 155 | ① | GUO等[ |
| 内含子4 | c.238+4T>C | 剪接突变 | 14 | 127(-4.27s) | 130 | ①+② | SHAW等[ |
| 外显子4 | C.239A>G | 错义突变 | 37 | 159(-2.25s) | ①+③ | LIN等[ | |
| 内含子4 | c.239-10_239-7delATTA | 剪接突变 | GEDEON等[ | ||||
| 内含子4 | c.239-11_239-4delAATTATTT | 剪接突变 | GEDEON等[ | ||||
| 内含子4 | c.239-11_239-9delAAT | 剪接突变 | 42 | 148(-4.03s) | ①+③ | DAVIS等[ | |
| 外显子5 | c.241_242delAT | 缺失突变 | GEDEON等[ | ||||
| 外显子5 | c.241_242delAT | 缺失突变 | 15 | ② | MUMM等[ | ||
| 外显子5 | c.248T>C | 错义突变 | GRUNEBAUM等[ | ||||
| 外显子5 | c.260A>C | 错义突变 | 13 | 126(-1.89s) | 138 | 无 | ZHANG等[ |
| 外显子 5 | c.262_266delGACAT | 缺失突变 | GEDEON等[ | ||||
| 外显子5 | c.262_266delGACAT | 缺失突变 | 25 | 143(-4.87s) | 154 | ③ | 孔祥东等[ |
| 外显子5 | c.267_271delAAGAC | 缺失突变 | 26 | 158(-2.41s) | 178 | ③ | FIEDLER等[ |
| 外显子5 | c.267_27ldelAAGAC | 缺失突变 | 10 | 120(-3.21s) | 无 | 李娟等[ | |
| 外显子 5 | c.271 C>T | 无义突变 | GEDEON等[ | ||||
| 外显子5 | c.271C>T | 无义突变 | 13 | 139(-2.73s) | 130 | 无 | 孟婕等[ |
| 外显子 5 | c.271-275delCAAGA | 缺失突变 | GEDEON等[ | ||||
| 外显子5 | c.271-275delCAAGA | 缺失突变 | 29 | 135 (-6.18s) | 无 | 黄欢等[ | |
| 外显子5 | C.271-275delCAAGA | 缺失突变 | 37 | 118(-8.97s) | ①+③ | 徐贵江等[ | |
| 外显子5 | C.271-275del | 缺失突变 | 11 | 141(-1.11s) | 147 | 无 | 本研究家系1 |
| 外显子5 | C.271-275del | 缺失突变 | 11 | 138(-1.14s) | 145 | ② | 本研究家系2 |
| 外显子 5 | c.272-273delAA | 缺失突变 | GEDEON等[ | ||||
| 外显子5 | c.293delT | 缺失突变 | 27 | 112(-9.95s) | 142 | ①+③ | XIAO等[ |
| 外显子5 | c.320dupT | 插入突变 | 16 | 147(-4.02s) | 164 | GEDEON等[ | |
| 内含子5 | c.325-2A>C | 剪接突变 | 成人 | 152(-3.39s) | 168 | ① | GEDEON等[ |
| 内含子5 | c.325-10_3254delTCTTTCCinsAA | 剪接突变 | GEDEON等[ | ||||
| 内含子5~ 外显子 6 | 1 330 bp片段缺失 | 缺失突变 | GEDEON等[ | ||||
| 内含子5~ 外显子 6 | 1 371~1 445 bp片段缺失 | 缺失突变 | CHRISTIE等[ | ||||
| 内含子5~ 外显子 6 | 750 bp片段缺失 | 缺失突变 | CHRISTIE等[ | ||||
| 内含子5~ 外显子 6 | c.325-2_335delAGTTTTCAATGAA | 缺失突变 | 16 | 143(-4.61s) | 151 | 无 | 麻宏伟等[ |
| 内含子5~ 外显子6 | 1.335 kb片段缺失 | 缺失突变 | 20 | 157(-2.57s) | 172 | ① | SHAW等[ |
| 外显子6 | 外显子6缺失 | 缺失突变 | 成人 | 153(-3.31s) | 171 | ③ | GEDEON等[ |
| 外显子6 | c.328delT | 缺失突变 | FIEDLER等[ | ||||
| 外显子6 | c.329C>A | 无义突变 | 16 | 143(-4.61s) | 153 | 无 | SHI等[ |
| 外显子6 | c.333_336delGAAT | 缺失突变 | 14 | 142(-3.32s) | ② | SHAW等[ | |
| 外显子6 | c.345_346delTG | 缺失突变 | FIEDLER等[ | ||||
| 外显子6 | c.370-371insA | 插入突变 | 20 | 153(-3.23s) | 167 | 无 | XIA等[ |
| 外显子6 | c.387delA | 缺失突变 | 16 | 147(-3.97s) | ① | BAR-YOSEF等[ | |
| 外显子6 | c. 389 T>A | 错义突变 | GEDEON等[ | ||||
| 外显子6 | c.391C>T | 插入突变 | 14 | 146(-2.76s) | 170 | 无 | TAKAHASHI等[ |
| 外显子6 | c.391_392del | 缺失突变 | 28 | ①+② | HE等[ | ||
| 外显子6 | c.406A>T | 错义突变 | 15 | 134(-5.44s) | 144 | ①+② | YASAR等[ |

图4 不同TRAPPC2基因型与SEDT患儿成年终身高的关系 注:不同TRAPPC2基因变异导致的SEDT患儿身高无明显差异。
| [1] | 张闻宇, 康可, 张玉薇, 等. X-连锁迟发性脊椎骨骺发育不良家系TRAPPC2基因新无义突变的鉴定及分析[J]. 中华骨科杂志, 2022, 42(5):313-319. |
| [2] |
GEDEON A K, TILLER G E, LE MERRER M, et al. The molecular basis of X-linked spondyloepiphyseal dysplasia tarda[J]. Am J Hum Genet, 2001, 68(6):1386-1397.
DOI PMID |
| [3] | 朱岷, 熊丰. 骨骺发育障碍与生长[J]. 中国实用儿科杂志, 2020, 35(6):455-460. |
| [4] | 刘宇, 王环环, 肖冰, 等. X连锁迟发性脊椎骨骺发育不良家系TRAPPC2基因缺失突变的高通量测序分析[J]. 上海交通大学学报(医学版), 2024, 44(3):407-411. |
| [5] |
WON J Y, KIM D, PARK S Y, et al. Novel loss-of-function variants of TRAPPC2 manifesting X-linked spondyloepiphyseal dysplasia tarda:report of two cases[J]. BMC Med Genet, 2019, 20:70.
DOI |
| [6] |
MILLER D T, LEE K, CHUNG W K, et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing:a policy statement of the American College of Medical Genetics and Genomics(ACMG)[J]. Genet Med, 2021, 23(8):1381-1390.
DOI URL |
| [7] | RICHARDS S, AZIZ N, BALE S, et al. Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5):405-424. |
| [8] | RIVERA-MU OZ E A, MILKO L V, HARRISON S M, et al. ClinGen Variant Curation Expert Panels:delivering a unified approach to variant interpretation[J]. Genet Med, 2020, 22(4):818-826. |
| [9] |
BRNICH S E, RIVERA-MU OZ E A, BERG J S. Clinical Genome Resource:standards for functional evidence evaluation[J]. Genet Med, 2019, 21(11):2412-2421.
DOI URL |
| [10] | MUMM S, CHRISTIE P T, FINNEGAN P, et al. A five-base pair deletion in the sedlin gene causes spondyloepiphyseal dysplasia tarda in a six-generation Arkansas kindred[J]. J Clin Endocrinol Metab, 2000, 85(9):3343-3347. |
| [11] | GEDEON A K, COLLEY A, JAMIESON R, et al. Identification of the gene(SEDL) causing X-linked spondyloepiphyseal dysplasia tarda[J]. Nat Genet, 1999, 22(4):400-404. |
| [12] |
LI Y, WU H, LI H. Growth hormone therapy in a boy with X-linked spondyloepiphyseal dysplasia tarda:a 3-year observation[J]. Endokrynol Pol, 2021, 72(4):410-411.
DOI URL |
| [13] | WANG H L, GAO C, LUO Q, et al. Gene diagnosis of X-linked spondyloepiphyseal dysplasia tarda by linkage analysis and DNA sequencing[J]. Zhonghua Er Ke Za Zhi, 2003, 41(4):256-259. |
| [14] |
MATSUI Y, YASUI N, OZONO K, et al. Loss of the SEDL gene product(Sedlin) causes X-linked spondyloepiphyseal dysplasia tarda:identification of a molecular defect in a Japanese family[J]. Am J Med Genet, 2001, 99(4):328-330.
DOI URL |
| [15] |
GOMES M E S, KANAZAWA T Y, RIBA F R, et al. Novel and recurrent mutations in the FGFR3 gene and double heterozygosity cases in a cohort of Brazilian patients with skeletal dysplasia[J]. Mol Syndromol, 2018, 9(2):92-99.
DOI URL |
| [16] |
XIA X Y, YU J, LI W W, et al. A novel nonsense mutation in the sedlin gene(SEDL) causes severe spondyloepiphyseal dysplasia tarda in a five-generation Chinese pedigree[J]. Genet Mol Res, 2014, 13(2):3362-3370.
DOI URL |
| [17] | LOU G, ZHAO Y, ZHAO H, et al. Functional analysis of a novel nonsense variant c.91A>T of the TRAPPC2 gene in a Chinese family with X-linked recessive autosomal spondyloepiphyseal dysplasia tarda[J]. Front Genet, 2023, 14:1216592. |
| [18] |
ADACHI H, TAKAHASHI I, TAKAHASHI T. Novel TRAPPC2 mutation in a boy with X-linked spondylo-epiphyseal dysplasia tarda[J]. Pediatr Int, 2014, 56(6):925-928.
DOI URL |
| [19] |
RYU H, PARK J, CHAE H, et al. X-linked spondyloepiphyseal dysplasia tarda:identification of a TRAPPC2 mutation in a Korean pedigree[J]. Ann Lab Med, 2012, 32(3):234-237.
DOI URL |
| [20] | WU X, DENG K, WANG C, et al. Mutation analysis of the TRAPPC2 gene in a Chinese family with X-linked spondyloepiphyseal dysplasia tarda[J]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 2015, 32(4):476-480. |
| [21] |
WANG H, WU W, XU Z, et al. A novel splicing mutation in the SEDL gene causes spondyloepiphyseal dysplasia tarda in a large Chinese pedigree[J]. Clin Chim Acta, 2013, 425:30-33.
DOI URL |
| [22] |
FUKUMA M, TAKAGI M, SHIMAZU T, et al. A familial case of spondyloepiphyseal dysplasia tarda caused by a novel splice site mutation in TRAPPC2[J]. Clin Pediatr Endocrinol, 2018, 27(3):193-196.
DOI URL |
| [23] | FIEDLER J, LE MERRER M, MORTIER G, et al. X-linked spondyloepiphyseal dysplasia tarda:novel and recurrent mutations in 13 European families[J]. Hum Mutat, 2004, 24(1):103. |
| [24] |
TAKAGI M, YAGI H, NAKAMURA Y, et al. A case of spondyloepiphyseal dysplasia tarda caused by a novel intragenic deletion of TRAPPC2[J]. Clin Pediatr Endocrinol, 2015, 24(3):139-141.
DOI URL |
| [25] | 黄颖, 谢杰, 郑诗瑶, 等. 中国南方一罕见迟发性脊椎骨骺发育不良大家系的表型和基因型的相关性研究[J]. 中山大学学报(医学科学版), 2022, 43(3):392-399. |
| [26] | 王莉莉, 吴海瑛, 孙辉, 等. X-连锁迟发性脊椎骨骺发育不良一家系TRAPPC2基因变异分析[J]. 临床儿科杂志, 2020, 38(5):321-323. |
| [27] |
YUNIS E, FONTALVO J, QUINTERO L. X-linked Dyggve-Melchior-Clausen syndrome[J]. Clin Genet, 1980, 18(4):284-290.
PMID |
| [28] | FIEDLER J, FRANCES A M, MERRER M L, et al. X-linked spondyloepiphyseal dysplasia tarda:molecular cause of a heritable platyspondyly[J]. Spine, 2003, 28(22):E478-E482. |
| [29] | 朱海燕, 李洁, 朱瑞芳, 等. X-连锁迟发性脊椎骨骺发育不良家系基因突变研究[J]. 中华医学遗传学杂志, 2008, 25(4):421-423. |
| [30] | CAO F, WANG Q W, YU B, et al. Prenatal diagnosis of a case with X-linked spondyloepiphyseal dysplasia tarda[J]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 2013, 30(5):598-600. |
| [31] | TAMHANKAR P M, KULKARNI A, VASUDEVAN L. X-linked spondyloepiphyseal dysplasia tarda with mutation in TRAPPC2 gene:first report from India[J]. J Orthop Case Rep, 2020, 10(2):1-4. |
| [32] | CHRISTIE P T, CURLEY A, NESBIT M A, et al. Mutational analysis in X-linked spondyloepiphyseal dysplasia tarda[J]. J Clin Endocrinol Metab, 2001, 86(7):3233-3236. |
| [33] |
ZHANG C, DU C, YE J, et al. A novel deletion variant in TRAPPC2 causes spondyloepiphyseal dysplasia tarda in a five-generation Chinese family[J]. BMC Med Genet, 2020, 21:117.
DOI PMID |
| [34] | 王莉, 姚丰, 廖世秀, 等. X-连锁迟发性脊髓骨骺发育不良家系的基因诊断[J]. 中华检验医学杂志, 2010, 33(6):527-530. |
| [35] | ZHOU W J, ZHOU S Y, HUANG S W, et al. Identification of a missense mutation in SEDL gene from a Chinese family with X-linked spondyloepiphyseal dysplasia tarda[J]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 2008, 25(1):15-18. |
| [36] |
GUO H, XU X, WANG K, et al. A novel RNA-splicing mutation in TRAPPC2 gene causing X-linked spondyloepiphyseal dysplasia tarda in a large Chinese family[J]. J Genet, 2009, 88(1):87-91.
DOI URL |
| [37] |
SHAW M A, BRUNETTI-PIERRI N, KADASI L, et al. Identification of three novel SEDL mutations,including mutation in the rare,non-canonical splice site of exon 4[J]. Clin Genet, 2003, 64(3):235-242.
DOI URL |
| [38] | LIN Y, RAO S Q, YANG Y. A novel mutation in the SEDL gene leading to X-linked spondyloepiphyseal dysplasia tarda in a large Chinese pedigree[J]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 2008, 25(2):150-153. |
| [39] |
DAVIS E E, SAVAGE J H, WILLER J R, et al. Whole exome sequencing and functional studies identify an intronic mutation in TRAPPC2 that causes SEDT[J]. Clin Genet, 2014, 85(4):359-364.
DOI URL |
| [40] |
GRUNEBAUM E, ARPAIA E, MACKENZIE J J, et al. A missense mutation in the SEDL gene results in delayed onset of X linked spondyloepiphyseal dysplasia in a large pedigree[J]. J Med Genet, 2001, 38(6):409-411.
DOI URL |
| [41] |
ZHANG L, WANG J, DONG G, et al. A novel missense variant in TRAPPC2 causes X-linked spondyloepiphyseal dysplasia tarda:a case report[J]. Medicine, 2021, 100(11):e25169.
DOI URL |
| [42] | 孔祥东, 刘宁, 史惠蓉, 等. X-连锁迟发性脊柱骨骺发育不良家系TRAPPC2基因突变分析[J]. 中华检验医学杂志, 2013, 36(7):634-637. |
| [43] | 李娟, 柴晓静, 陆莉, 等. X连锁迟发性脊椎骨骺发育不良SEDL基因突变的分析[J]. 中华医学遗传学杂志, 2014, 31(5):604-607. |
| [44] | 孟婕, 彭慧芳, 姜志红. X-连锁迟发性脊椎骨骺发育不良1例[J]. 中国临床案例成果数据库, 2022, 4(1):E06111. |
| [45] | 黄欢, 王珏, 洪蕾, 等. X连锁迟发性脊椎骨骺发育不良家系的基因检测及其产前诊断[J]. 中华妇产科杂志, 2015, 50(8):614-616. |
| [46] | 徐贵江, 李茜, 刘明艳, 等. 1个X-连锁迟发性脊椎骨骺发育不良家系的TRAPPC2基因突变分析[J]. 临床检验杂志, 2018, 36(5):396-398. |
| [47] | XIAO C, ZHANG S, WANG J, et al. A single nucleotide deletion of 293delT in SEDL gene causing spondyloepiphyseal dysplasia tarda in a four-generation Chinese family[J]. Mutat Res, 2003, 525(1-2):61-65. |
| [48] | 麻宏伟, 姜俊, 吕峻峰, 等. X-连锁迟发性脊椎骨骺发育不良SEDL基因剪接受体突变导致潜在剪接位点激活[J]. 中华医学遗传学杂志, 2005, 22(3):251-253. |
| [49] |
SHI Y R, LEE C C, HSU Y A, et al. A novel nonsense mutation of the sedlin gene in a family with spondyloepiphyseal dysplasia tarda[J]. Hum Hered, 2002, 54(1):54-56.
DOI URL |
| [50] |
XIA X Y, CUI Y X, ZHOU Y C, et al. A novel insertion mutation in the SEDL gene results in X-linked spondyloepiphyseal dysplasia tarda in a large Chinese pedigree[J]. Clin Chim Acta, 2009, 410(1-2):39-42.
DOI URL |
| [51] |
BAR-YOSEF U, OHANA E, HERSHKOVITZ E, et al. X-linked spondyloepiphyseal dysplasia tarda:a novel SEDL mutation in a Jewish Ashkenazi family and clinical intervention considerations[J]. Am J Med Genet A, 2004, 125A(1):45-48.
DOI URL |
| [52] |
TAKAHASHI T, TAKAHASHI I, TSUCHIDA S, et al. An SEDL gene mutation in a Japanese kindred of X-linked spondyloepiphyseal dysplasia tarda[J]. Clin Genet, 2002, 61(4):319-320.
DOI URL |
| [53] | HE Z, DAI S M, CHEN Z. Clinical images:back pain,flattened vertebral bodies,and a novel mutation in the TRAPPC2 gene[J]. ACR Open Rheumatol, 2025, 7(2):e70000. |
| [54] |
YASAR D, G LERAY LAFCI N, KARACAN K KALI G, et al. A novel premature termination codon mutation in TRAPPC2 is associated with X-linked spondyloepiphyseal dysplasia tarda[J]. Mol Syndromol, 2025, 16(5):461-468.
DOI URL |
| [55] |
GREER S Y, BULLION E A. Reconstruction of anatomy and care provisioning in a severe case of spondyloepiphyseal dysplasia[J]. Int J Paleopathol, 2021, 34:147-154.
DOI PMID |
| [56] |
马端. 提升罕见遗传疾病病因阳性检出率的思考及应用[J]. 检验医学, 2025, 40(2):109-113.
DOI |
| [1] | 李仁强, 罗俊峰, 陈云弟. α-地中海贫血和β-地中海贫血基因突变异尾双链荧光探针杂交法的建立及应用评价[J]. 检验医学, 2022, 37(10): 955-962. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
