{kind=link}
晚期糖基化终产物与冠心病发病机制的研究进展
引用本文
刘军锋, 孔美娟, 贾克刚. 晚期糖基化终产物与冠心病发病机制的研究进展. 检验医学, 2014, 29(1): 76-80[LIU Junfeng, KONG Meijuan, JIA Kegang. Research on advanced glycation end products in the mechanism of coronary artery disease. Labratory Medicine, 2014, 29(1): 76-80] 
Permissions
晚期糖基化终产物与冠心病发病机制的研究进展
摘要
晚期糖基化终产物 (AGEs) 密切参与了血管平滑肌细胞 (VSMCs) 分化与增殖以及冠心病等心血管疾病的病理生理过程。AGEs能通过单核细胞趋化蛋白 (ERK) 、丝氨酸/苏氨酸蛋白激酶 (Akt) 信号通路来诱导VSMCs的自噬作用, 可依赖骨髓基质细胞衍生因子-1 (SDF-1)/趋化因子受体CXCR4轴信号通路促进心肌微血管内皮细胞 (CMECs) 的增生。AGEs-2和AGEs-3上调了单核细胞AGEs受体 (RAGE) 的表达。AGEs能抑制内皮祖细胞 (EPCs) 的增殖、迁移和黏附功能并诱导EPCs凋亡;能增加平滑肌细胞结缔组织因子 (CTGF) mRNA和蛋白质的表达, 刺激心肌成纤维细胞增殖并分泌转化生长因子-β1 (TGF-β1),同时诱导Smad2及Smad4的表达。羧甲基赖氨酸 (CML)/RAGE轴通过主动脉平滑肌成骨细胞的分化, 诱导巨噬细胞凋亡, 从而使AGEs在糖尿病动脉粥样硬化中发挥重要作用。可溶性RAGE (sRAGE) 可作为RAGE配体的诱饵来防止动脉粥样硬化, 其灵敏度和阴性预测值在判定冠状动脉介入治疗 (PCI) 术后再狭窄方面均高于AGEs/ sRAGE比值。ALT-711是一种AGEs的裂解剂, 能明显抑制AGEs介导的活性氧 (ROS) 产生、细胞外信号调节激酶磷酸化及环氧合酶-2的表达;色素上皮衍生因子 (PEDF) 能抑制AGEs诱导的血小板CD40配体 (CD40L) 表达, 从而有可能成为预防冠心病的一个治疗靶点。他汀类药物亦能抑制AGEs诱导主动脉平滑肌细胞的增殖及ROS的产生。通过以上诸多因素的研究, 可揭示冠心病的某些发病机制, 为相应干预药物的研究及调整临床治疗策略提供依据和方向。
关键词:
晚期糖基化终产物; 晚期糖基化终产物受体; 冠心病; 动脉粥样硬化; 血管平滑肌细胞; 信号传导
Research on advanced glycation end products in the mechanism of coronary artery disease
Abstract
Advanced glycation end products (AGEs) are closely involved in the pathophysiological process of vascular smooth muscle cell (VSMCs) differentiation, proliferation, coronary artery disease and other cardiovascular diseases. AGEs can induce autophagy in VSMCs through the extracellular signal-regulated protein kinase (ERK) and serine-threonine kinase (Akt) signaling pathways. AGEs improve cardiac microvascular endothelial cell (CMECs) proliferation depending on bone marrow stromal cell-derived factor-1 (SDF-1)/ chemokine receptor CXCR4 axis signaling pathway. AGEs-2 and AGEs-3 up-regulate the expression of receptor for advanced glycation end product (RAGE) on monocytes. AGEs inhibit the proliferation, migration and adhesion of endothelial progenitor cell (EPCs), and induce the apoptosis of EPCs. AGEs increase the expression activity of connective tissue growth factor (CTGF) mRNA and protein, and promote the proliferation of cardiac fibroblast and the secretion of transforming growth factor-beta 1 (TGF-β1) and induce the expressions of Smad2 and Smad4. The carboxymethyl lysine (CML)/RAGE axis plays an important role in atherosclerotic calcification of diabetes through the mechanism that induces the apoptosis of macrophages followed by the osteogenic differentiation of aortic smooth muscle cells. Soluble form of RAGE (sRAGE) can be served as bait of RAGE ligand to prevent atherosclerosis, and the sensitivity and negative predictive value of sRAGE are higher than those of AGEs/sRAGE ratio in identifying post-percutaneous coronary intervention (PCI) restenosis. ALT-711 is a breaker of AGEs-based cross links, which can inhibit AGEs-mediated formation of reactive oxygen species (ROS), extracellular signal-regulated kinase phosphorylation and cyclooxygenase-2 expression. Pigment epithelium-derived factor (PEDF) can inhibit CD40 ligand (CD40L) overexpression by blocking the effects of AGEs on platelets, which may become a therapeutic target for the prevention of coronary artery disease. Statins can also inhibit AGEs-induced aortic smooth muscle cell proliferation and production of ROS. The research above can reveal some the pathogenesis of coronary artery disease, provide foundation and direction for exploring the corresponding drug intervention and adjustment of clinical treatment strategies.
Keyword:
Advanced glycation end product; Receptor for advanced glycation end product; Coronary artery disease; Atherosclerosis; Vascular smooth muscle cell; Signal transduction
晚期糖基化终产物 (advanced glycation end products, AGEs) 代表了一群异质性的蛋白质、脂类、核酸与还原糖不可逆的交叉连接产物。AGEs可通过刺激炎症反应、促进动脉粥样硬化斑块形成、调节血管硬度, 密切参与冠心病的病理生理过程[ 1]。AGEs可通过AGEs受体 (receptor for advanced glycation end products, RAGE) 和非受体途径从多个方面参与了心肌损害的过程, AGEs可通过血小板衍生因子释放、血管平滑肌细胞 (vascular smooth muscle cells, VSMCs) 内信号传递途径的变化、内皮细胞结构和功能改变, 从而使VSMCs分化与增殖并导致血管增厚, 促进动脉粥样硬化并影响血流动力, 参与心血管的功能调节和结构重塑, 最终导致冠心病的发生。但AGEs对冠心病诱导的调节机制仍未完全清楚。我们结合近年文献对这一领域的热点研究进行综述。
一、AGEs对细胞信号传导的影响
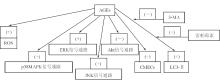
自噬作用是一种维持生命的过程, 在动脉粥样斑块中通过脂质氧化、炎症、代谢应激, 使这种作用加强。Hu等[ 2]的研究结果表明, 用AGEs处理的大鼠VSMCs活化了细胞外信号调节蛋白激酶 (extracellular signal-regulated protein kinase, ERK) 、c-Jun氨基末端激酶 (c-Jun NH2-terminal kinase, JNK) 、p38丝裂原活化蛋白激酶 (p38 mitogen-activated protein kinase, p38MAPK) 的信号通路, 但抑制了丝氨酸/苏氨酸蛋白激酶 (serine-threonine kinase, Akt) 的通路。用ERK抑制剂和Akt活化剂预处理能抑制AGEs诱导自噬作用的发生, 显示AGEs能通过ERK和Akt信号通路来诱导VSMCs的自噬作用, 从而有助于VSMCs增殖的进程, 与糖尿病动脉粥样硬化相关联。Xie等[ 3]应用AGEs修饰的牛血清白蛋白 (AGEs-modified bovine serum albumin, AGEs-BSA) 诱导上调了微管相关蛋白1轻链3-Ⅱ (light chain 3-Ⅱ, LC3-Ⅱ) 的水平。这种诱导的调节作用可被自噬抑制剂3-甲基腺嘌呤 (3-methyladenine, 3-MA) 所抑制, 但可被自噬诱导剂雷帕霉素所刺激。在AGEs-BSA诱导下乳酸脱氢酶外泄露增加, 3-MA可提高此种效果, 但雷帕霉素没有此作用。α-维生素E是一种氧化酶抑制剂, 不仅能降低由AGEs-BSA诱导所增加的活性氧 (reactive oxygen species, ROS),而且还能降低上调的LC3-Ⅱ蛋白水平。见图1。
粟妍晖等[ 4]分离的心肌微血管内皮细胞 (cardiac microvascular endothelial cells, CMECs) 在AGEs的作用下, 增殖能力和营养结构形成能力显著增强, 并可表达CXC趋化因子受体-4 (CXC chemokine receptor type 4, CXCR4 ) 和骨髓基质细胞衍生因子-1 (stromal cell-derived factor 1, SDF-1),SDF-1能诱导CMECs的增殖和管样结构形成, CXCR4阻滞剂AMD3100能抑制SDF-1的诱导作用。实验结果表明, AGEs可通过SDF-1/ CXCR4轴信号通路促进CMECs的增殖, 并诱导细胞管样结构的形成。见图1:
 | 图1 AGEs对某些信号传导途径的影响 |
Takahashi等[ 5]研究了4种不同的AGEs亚型 (AGEs-2、AGEs-3、AGEs-4、AGEs-5) 在0.1~100.0 μg/mL范围内对细胞内黏附分子-1、B7.1、B7.2和单核细胞CD40的表达影响及其对人外周血单核细胞γ-干扰素、肿瘤坏死因子-α (tumor necrosis factor alpha, TNF-α) 的影响。AGEs-2和AGEs-3能有选择的诱导黏附分子的表达和细胞因子的产生。使用黏附分子的拮抗实验表明, 单核细胞和T/自然杀伤细胞之间相互作用参与了AGEs-2和AGEs-3诱导细胞因子产生的过程, AGEs-2和AGEs-3上调了单核细胞RAGE的表达, 核转录因子-κB (nuclear transcription factor kappa B, NF-κB) 和p38MAPK抑制剂可抑制AGEs-2和AGEs-3的作用。糖基化血清白蛋白 (glycation serum albumin, GSA) 是AGEs中重要的一种。GSA可以促进VSMCs单核细胞趋化蛋白-1 (monocyte chemotactic protein 1, MCP-1) 和白细胞介素-8 (interleukin 8, IL-8) 的表达。这可能是通过激活细胞内p38MAPK信号转导通路, 促进NF-κB的活化而实现的[ 6]。
二、AGEs对血管细胞、心肌细胞及单核细胞的影响
内皮祖细胞 (endothelial progenitor cells, EPCs) 在防止动脉粥样硬化起了重要作用, 但调节EPCs功能的因素还不完全清楚。内皮细胞的损伤在冠心病发病机制中发挥了一定的作用[ 7]。AGEs通过促进血管内皮细胞的增殖、迁移、损伤和死亡, 从而成为糖尿病和动脉粥样硬化发病机制的一部分。Ueda等[ 8]的研究结果显示, AGEs的血清水平和吸烟与低水平的EPCs数量呈独立相关。此外, 在女性中, AGEs和低水平的高密度脂蛋白胆固醇与低水平的EPCs迁移活性呈独立相关。Chen等[ 9]在含有AGEs成份的环境中培养EPCs, 结果显示EPCs生成ROS增加、抗氧化剂的防御能力降低、氧化应激能力增强, 从而使EPCs增殖、迁移和黏附功能受到抑制并诱导EPCs凋亡。
Nam等[ 10]将单核细胞、内皮细胞、平滑肌细胞在100 μg/mL AGEs乙醇醛 (glycolaldehyde-induced, glycol-AGEs) 中共培养24~48 h, 结果显示glycol-AGEs诱导平滑肌细胞增殖, 同时平滑肌细胞也表达白细胞介素-6 (interleukin-6, IL-6) 和MCP-1, 且在含有glycol-AGEs的共培养系统中细胞因子的表达也明显升高。在含有AGEs和血清白蛋白的环境中对人单核细胞进行培养, CD36表达水平和细胞内ROS明显增加[ 11]。AGEs能增加平滑肌细胞结缔组织生长因子 (connective tissue growth factor, CTGF) mRNA和蛋白质的表达, 且这种增加效应具有时间和浓度依赖性, 提示AGEs能通过该途径促进动脉粥样硬化的发生、发展[ 12]。李洁等[ 13]的研究结果表明AGEs能刺激心肌成纤维细胞增殖并分泌转化生长因子-β1 (transforming growth factor-β1, TGF-β1),同时诱导CTGF、Smad2及Smad4的表达。
三、RAGE与动脉粥样硬化
RAGE是AGEs的主要信号转导受体。此外, RAGE也是一个多配体结合的免疫球蛋白超家族成员。除了AGEs, RAGE还结合了S100/钙粒蛋白家族、高机动性组框1、β-淀粉样肽和β-折叠纤维中的某些成员[ 14]。AGEs直接或通过受体介导的机制参与动脉粥样硬化的过程。目前, 研究最广泛的受体是RAGE。AGEs-RAGE的相互作用改变了细胞的信号传导, 促进了基因的表达, 增强了促炎症分子的释放, 引起众多不同类型的氧化应激反应[ 15]。在动物模型中, 中断AGEs-RAGE的相互作用可减轻病灶的面积和斑块的发展。在RAGE(-/-)/载脂蛋白E(-/-)双基因敲除的老鼠中, RAGE缺乏, 减轻了糖尿病动脉粥样硬化的发展[ 16]。
Wang等[ 17]通过实验表明, 小鼠主动脉壁上的羧甲基赖氨酸 (carboxymethyl lysine, CML) 沉积信号和RAGE表达主要限于动脉粥样硬化斑块部位。CML/RAGR轴可能首先启动了动脉粥样硬化损伤区域巨噬细胞的凋亡, 然后在高血脂、细胞凋亡并存的环境下诱导了骨形态生成蛋白-2 -核结合因子α1-碱性磷酸酶-动脉硬化的级联反应。作者认为CML/RAGE可能在糖尿病动脉粥样硬化中发挥了重要的作用。这种作用是通过主动脉平滑肌的成骨细胞分化, 诱导巨噬细胞凋亡来实现的。
通过RAGE介导的氧化应激能损害EPCs的功能, 并增加了氧化应激介导EPCs凋亡的敏感性。应用小干扰RNA (small interfering RNA, siRNA) 阻断RAGE的蛋白表达可减弱AGEs与EPCs共培养时对EPCs造成的影响[ 9]。利用含有AGEs和牛血清白蛋白的环境对人单核细胞进行培养, 并用siRNA阻止RAGE的表达, 结果表明CD36及ROS表达明显受到抑制[ 11]。
AGEs与其受体的相互作用增加了炎症介质和可溶性血管细胞黏附分子-1 (soluble vascular cell adhesion molecule 1, sVCAM-1) 的表达, 并诱导与动脉粥样硬化相关联的氧自由基表达。球囊损伤诱导动脉粥样硬化与AGEs和RAGE的表达增加有关。可溶性RAGE (soluble RAGE, sRAGE) 可作为RAGE配体的诱饵来防止动脉粥样硬化。McNair等[ 18]观察了46例非ST段抬高的心肌梗死患者进行冠状动脉介入治疗后sRAGE的水平变化, 除了19例术后再狭窄者外, 其余患者术前和术后sRAGE、TNF-α水平是相似的;再狭窄的患者术后sRAGE水平比术前低, 而TNF-α术后比术前高;sRAGE的灵敏度和阴性预测值高达100%,在判定冠状动脉介入治疗术后再狭窄方面均高于AGEs/sRAGE比值。
四、药物干预对AGEs的影响
ALT-711是一种AGEs交叉联接裂解剂。Kim等[ 19]探讨了ALT-711抑制AGEs诱导的效果。在ALT-711治疗中, 大鼠动脉平滑肌细胞的增生被明显抑制, ALT-711呈剂量依赖性的抑制了AGEs介导的ROS生成、细胞外信号调节激酶磷酸化、环氧合酶-2的表达。AGEs诱导结缔组织表达和细胞外基质表达的机制在ALT-711治疗中被减弱。体内研究显示, 用ALT-711治疗球囊损伤的大鼠, RAGE表达和内膜增生被明显抑制。但ALT-711是一种与硫胺素同源的结构, 硫胺素可被硫胺二磷酸激酶 (thiamine diphosphokinase, TDPK) 转化为硫胺素二磷酸 (thiamine diphosphate, TDP),而TDP是丙酮酸脱氢酶、α-酮戊二酸脱氢酶以及转酮酶的辅酶。这些酶活性的降低可由于TDP缺乏从而导致相关疾病。Krautwald等[ 20]研究了ALT-711是否为TDPK的抑制剂, 结果表明ALT-711是一种低亲和力的TDPK抑制剂, 其在治疗浓度时不会干扰硫胺素的代谢。但作者指出, 不应该再有新的、比ALT-711更具潜在竞争力的基于硫胺素设计的AGEs交叉联接蛋白裂解剂问世。
抑制血小板CD40配体 (CD40 ligand, CD40L) 的表达对预防冠心病来说可能是一个新的治疗靶点, 但色素上皮衍生因子 (pigment epithelium derived factor, PEDF) 对血小板CD40L表达的影响机制仍有待阐明。Yamagishi等[ 21]发现PEDF能通过减弱AGEs的影响, 从而起到抵抗心血管疾病的保护作用;与对照组相比, 糖尿病大鼠血小板CD40L的表达水平大约增加了2倍;通过使用PEDF治疗, CD40L的表达可部分被抑制;将AGEs注射入非糖尿病大鼠体内, CD40L的表达水平大约增加了1.3倍;通过应用PEDF治疗, 这种影响可完全被预防。因此PEDF的替代疗法可能会提供一种新的、有前途的预防糖尿病心血管疾病的途径。
Yoon等[ 22]研究表明, AGEs浓度的增多与大鼠主动脉平滑肌细胞 (aortic smooth muscle cell of sprague-dawley rat, RASMC) 增殖、ROS生成增多相关。RASMC增殖下降、ROS生成减少与使用的他汀类药物治疗呈剂量依赖性。AGEs能增加NF-κB P65、磷酸化ERK、磷酸化p38MAPK、环氧合酶-2、c-Jun的表达, 但这些物质的表达可被他汀类药物所抑制。糖尿病大鼠球囊损伤后新生内膜比假手术组增厚许多, 但被球囊损伤后经他汀类药物治疗很少有内膜增厚。有研究[ 23]显示阿托伐他汀可通过人内皮细胞过氧化物酶增殖物活化受体-γ (peroxisome proliferators-activated receptor gamma, PPAR-γ) 表达抑制AGEs诱导人内皮细胞炎症反应。此作用可能是通过PPAR-γ对NF-κB信号途径的抑制作用实现的。罗格列酮可作为干预药物抑制AGEs促纤维化的病理过程。影响Smad蛋白的效应可能是罗格列酮干扰TGF-β1/Smad信号传递的重要途径[ 13]。
五、结语
AGEs与糖尿病并发心血管疾病的高风险相关, 包括冠状动脉疾病和外周动脉疾病, 这就解释了糖尿病患者发病和死亡的大部分原因。AGEs和患冠心病风险的联系不断扩展, 从AGEs对细胞信号传递的影响、效应细胞结构及功能受损到AGEs-RAGE相互作用所致的病理损害, 揭示了VSMCs分化与增殖、冠心病的某些发病机制。基于AGEs及其受体RAGE在冠心病进展中所起的重要作用, 相应的干预药物需得到一定的发展。因此抗AGEs、抗RAGE的药物进一步研发及其在临床治疗策略上的调整将有望成为防治冠心病的新方向。
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|