
{kind=link}
{kind=link}
{kind=link}
一例血色病患者及其家系铁代谢调节基因的突变分析
引用本文
管宇, 安鹏, 张晓峰. 一例血色病患者及其家系铁代谢调节基因的突变分析. 检验医学2014, 29(6):679-684
GUAN Yu, AN Peng, ZHANG Xiaofeng. Mutation analysis of iron metabolism regulated genes in a patient of hemochromatosis. Labratory Medicine, 2014, 29(6):679-684
Permissions
GUAN Yu, AN Peng, ZHANG Xiaofeng. Mutation analysis of iron metabolism regulated genes in a patient of hemochromatosis. Labratory Medicine, 2014, 29(6):679-684
Copyright©2014, 《检验医学》编辑部
《检验医学》编辑部所有
一例血色病患者及其家系铁代谢调节基因的突变分析
通讯作者:张晓峰,联系电话:021-56639828-4520
作者简介:管 宇,女,1976年生,博士, 主管技师, 主要研究方向为血色病分子诊断。
摘要
目的
对1名临床诊断怀疑为原发性血色病的患者及家属进行血色病相关基因检测。
方法记录患者临床资料,采集患者及家属外周血,检测血清铁(SI)、总铁结合力(TIBC)、铁蛋白(SF)和转铁蛋白饱和度(TS);提取血液基因组DNA,采用聚合酶链反应(PCR)扩增铁代谢调节基因
患者SF、SI、TS明显升高。
关键词:
遗传性血色病; 铁过载; 转铁蛋白受体2
中图分类号:R596
文献标志码:A
文章编号:1673-8640(2014)06-0679-06
Mutation analysis of iron metabolism regulated genes in a patient of hemochromatosis
Abstract
Objective
To detect the mutation of hemochromatosis-related gene in a patient suspected with hereditary hemochromatosis and his family members.
MethodsThe peripheral blood samples were collected from the patient and his family members after recording patient's clinical data. The indices of iron metabolism including serum iron (SI), total iron binding capacity (TIBC), serum ferritin (SF)and transferrin saturation (TS) were determined. The genomic DNA from peripheral blood was isolated, and polymerase chain reaction (PCR) was used to amplify exon and intron splice junctions, and the 5' and 3' untranslated regions (UTRs) of
SF, SI and TS increased significantly in the patient. Mutation analysis in the patient revealed c.224C>T (p.Ala75Val) and c.714C>G (p.Ile238Met) heterozygous mutations respectively located in exon2 and exon5 of
Keyword:
Hereditary hemochromatosis; Iron overload; Transferrin receptor 2
血色病是体内铁过量沉积导致组织结构破坏和器官功能损害的疾病状态。按病因分为原发性和继发性两大类。继发性血色病多见于骨髓无效造血(如再生障碍性贫血、地中海贫血、骨髓异常增生综合征等)或铁摄入过多(如反复大量输血、注射铁剂等)。
原发性血色病又称为遗传性血色病(hereditary hemochromatosis,HH),是一种铁代谢紊乱性遗传病,常见于北欧人群。因铁代谢调节基因突变导致胃肠铁吸收增加,过多的铁逐渐沉积在肝、心、胰等组织实质细胞,诱导氧化自由基的形成,破坏细胞内部结构,导致器官功能障碍和衰竭,甚至引起死亡。HH常见的临床表现包括肝纤维化、肝硬化、肝癌、糖尿病、扩张型心肌病、心力衰竭、垂体及性腺功能减退、关节痛和皮肤色素沉着等症状。在线人类孟德尔遗传数据库(Online Mendelian Inheritance in Man,OMIM)根据不同基因突变情况将HH分为4个类型: HFE (1型,HLA-linked hemochromatosis)、 HJV(2A型,hemojuvelin)、 HAMP(2B型,hepcidin antimicrobial peptide)、 TFR2(3型,transferrin receptor 2)和 SLC40 A1(4型,feroportin)。其中, HFE基因突变是北欧HH最常见的病理类型。
在我国,HH较罕见,临床误诊、漏诊率较高,遗传学研究基础薄弱。因此,我们希望通过本研究引起医务工作者对HH早期诊断和遗传学研究的重视。
材料和方法
一、病例资料
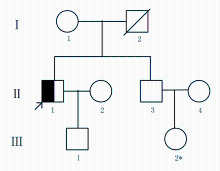
患者,男,52岁,汉族(见图1中Ⅱ:1)。因乏力、头昏、腹胀、2型糖尿病、扩张型心肌病入院。患者10年前出现性功能减退和皮肤色素沉着,未予重视。1年前确诊为糖尿病,予以胰岛素皮下注射,血糖控制不详。吸烟20年,约20支/d,无饮酒史,无溶血性疾病及输血史。否认家族遗传病史及传染病史。父亲死于肺气肿,其弟双手大关节疼痛病史10年,高血压1年,性功能正常,无肝病、心脏病及皮肤色素沉着。入院查体:面色青灰,中重度贫血貌。心界向左扩大,心尖部可闻及2~6级收缩期杂音。腹膨隆,移动性浊音阳性,双下肢凹陷性水肿。实验室检查:血红蛋白(hemoglobin,Hb) 103 g/L↓,随机血糖8.0 mmol/L,空腹血糖6.5 mmol/L,餐后2 h血糖10.6 mmol/L,γ-谷氨酰基转移酶(gamma-glutam-yltransferase,GGT) 52.08 U/L↑,总蛋白(total protein,TP) 58.95 g/L↓,白蛋白(albumin,Alb) 29.97 g/L↓,白/球比值(A/G) 1.061↓, 丙氨酸氨基转移酶(alanine aminotransferase,ALT) 56.07 U/L↑,天门冬氨酸氨基转移酶(aspartate aminotransferase,AST) 70.49 U/L↑,高敏肌钙蛋白T(high sensitivity troponin T,hs-cTnT)0.005 ng/mL↑,铁蛋白 1 697 ng/mL↑。甲、乙、丙型肝病毒检测阴性,溶血性贫血相关试验无异常。骨髓细胞学检查呈增生性贫血表现,细胞内铁阳性,细胞外铁++++。上腹部B超示:肝脏增大,肝硬化征象,脾脏增大,脾静脉增宽,腹腔大量积液。予腹穿抽水,腹水检查:腺苷脱氨酶(adenosine deaminase,ADA)28.15 U/L,抗酸杆菌阴性,未见恶性细胞。经会诊,排除继发性因素,考虑HH可能,需进一步做肝脏MRT平扫+增强及肝穿刺活检。但患者不能耐受肝脏MRT检查,拒绝做肝穿刺活检,同意进行遗传学检测。基因检测1个月后,患者死于心肌梗死。
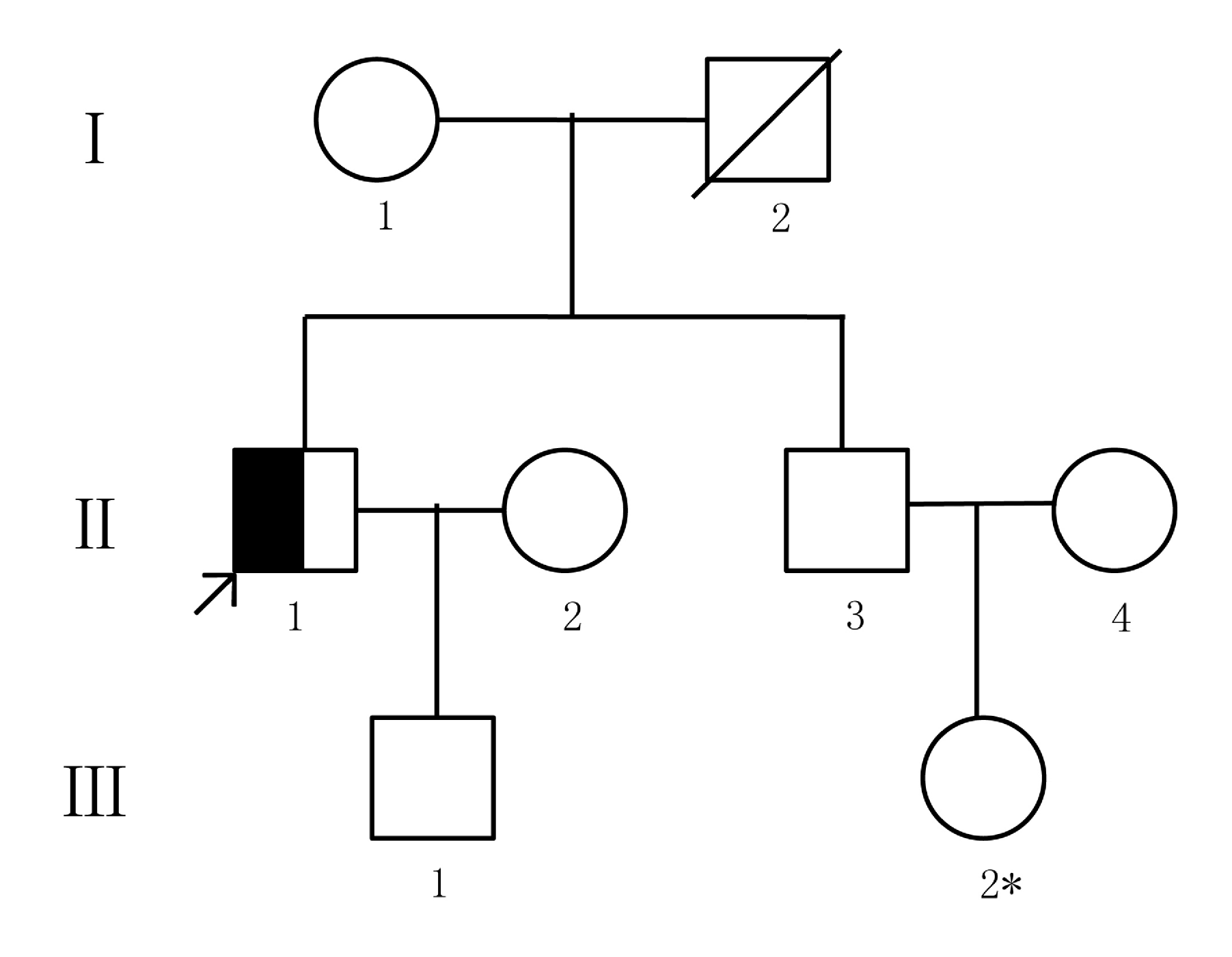 | 图 1. 患者家系图 |
二、血样采集
在告知并签署知情同意书后,采集患者及家属成员(共4 人)外周静脉血4 mL(抗凝管、促凝管各采集血液2 mL),1 500×g离心10 min,分离血清、血浆。 样本保存于-80 ℃冰箱。
三、铁代谢指标检测
采用电化学发光免疫分析法检测血清铁蛋白(serum ferritin,SF)浓度,仪器为罗氏cobas e601电化学发光免疫分析仪。采用化学比色法检测血清铁(erum iron,SI)、总铁结合力(total iron binding capacity,TIBC),仪器为日立7180型全自动生化分析仪。转铁蛋白饱和度(transferrin saturation,TS)=SI/TIBC×100%。参考区间:SF 男性 30~350 ng/mL,女性 15~300 ng/mL;SI男性 8.1~28.3 umol/L,女性 6.6~26 μmol/L;TIBC 46.4~69.5 μmol/L;TS 20%~55%。
四、基因检测
抽提患者及家属外周血基因组DNA(TIANGEN公司)。聚合酶链反应(polymerase chain reaction, PCR)扩增 HFE、HJV、HAMP、TFR2、 SLC40 A1基因外显子、内含子剪切序列及5'、3'端的UTR区域。扩增引物根据http://asia.ensembl.org/数据库设计,具体序列见表1。
| 表1 HFE/HJV/HAMP/TFR2 /SLC40 A1基因引物序列 |
PCR程序:95 ℃预变性5 min,95 ℃变性45 s,退火温度55 ℃ 45 s,72 ℃延伸90 s,35个循环。PCR产物经10 g/L琼脂糖电泳、切胶、回收(DNA gel extraction 试剂盒,德国QIAGEN公司),纯化产物送北京六合华大基因科技有限公司上海分公司双向直接测序。
结果
二、基因检测结果
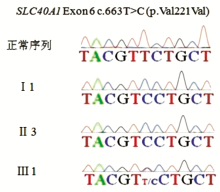
采用Lasergene SeqMan软件比对测序结果,发现患者 TFR2基因中出现两个杂合错义突变:exon2中第224位核苷酸胞嘧啶C突变为胸腺嘧啶T(224C>T),导致蛋白质中第75位丙氨酸(Ala)被缬氨酸(Val)取代(A75V,图2A);exon5中第714位胞嘧啶C突变为鸟嘌呤G(714C>G),导致第238位异亮氨酸(Ile)被甲硫氨酸(Met)取代(I238M,图2B)。 SLC40 A1基因第6个外显子上出现c.663T>C纯合突变(p.Val221Val同义突变,图2C)。 HFE、HAMP、HJV基因未见突变。
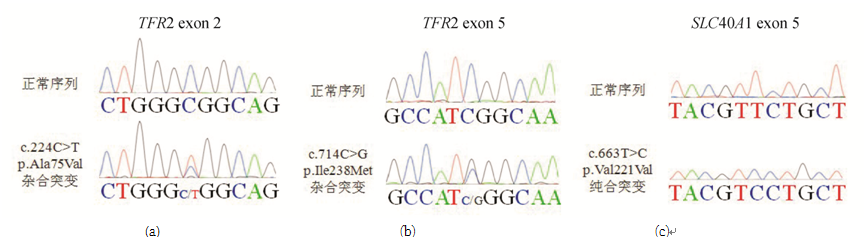 | 图2 患者 TFR2和 SLC40 A1基因突变结果注:(a) TFR2 exon 2中C>T杂合突变;(b) TFR2 exon 5中C>G杂合突变;(c) SLC40 A1 exon 6中T>C纯合突变 |
三、家系成员检测结果
检测患者母亲、弟弟、儿子外周血基因组DNA中上述突变位点,未发现 TFR2 A75V或I238M突变,皆存在 SLC40 A1 V221V同义突变(图3)。
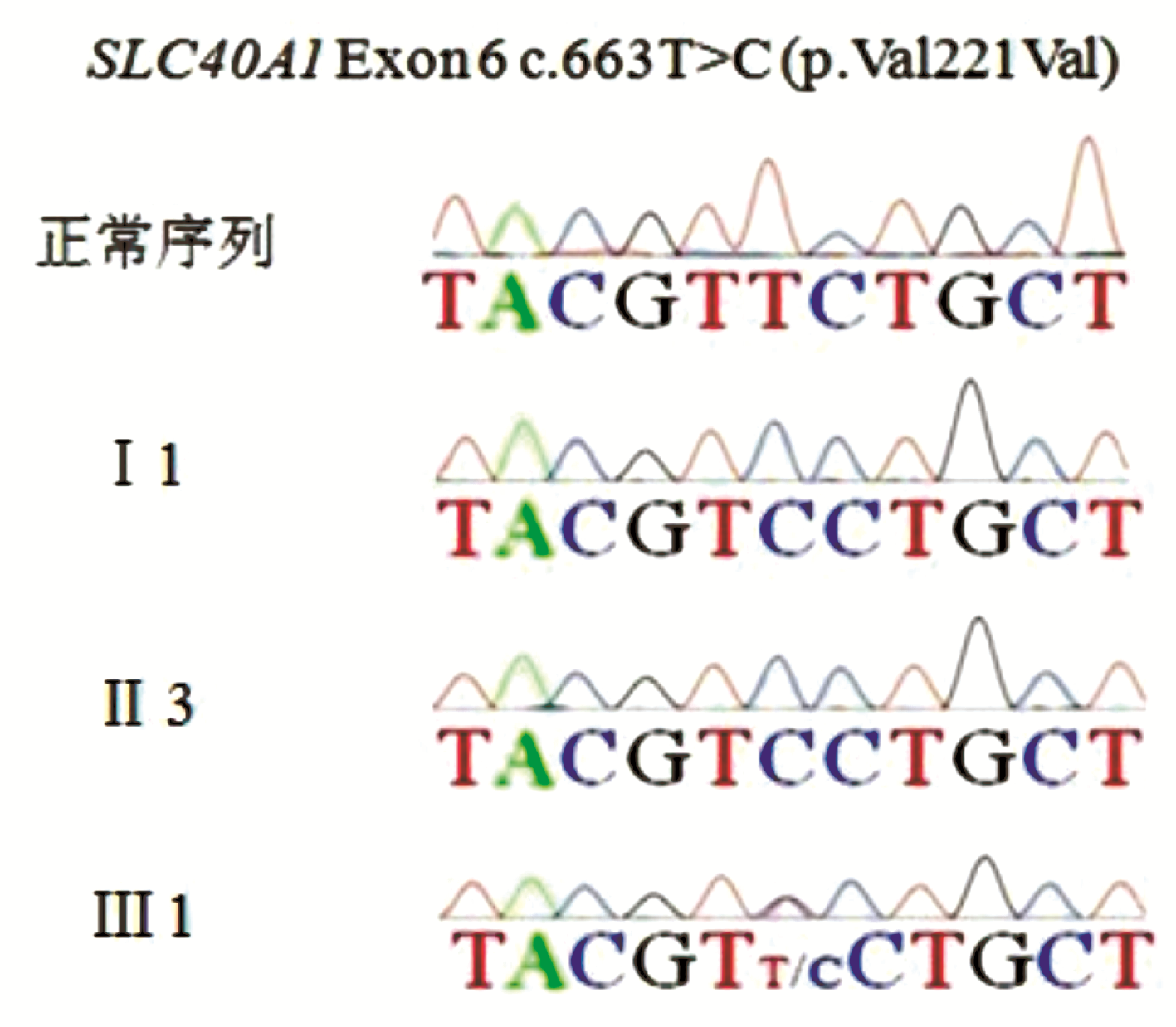 | 图3 家属成员SLC40A1基因突变结果 |
讨论
HFE是第一个被确定的血色病相关基因, 其中C282Y(酪氨酸Tyr取代282位半胱氨酸Cys)和H63D(天冬氨酸Asp取代63位的组氨酸His)是最主要的两个致病性突变位点。在北欧,大约85%~90%的HH患者带有C282Y纯合突变,5%为C282Y/H63D复合突变。但是 HFE C282Y在亚洲人群中的等位基因频率较低。这提示亚洲人群的HH遗传背景与欧美不同,可能以非 HFE相关性血色病(non- HFE related HH)为主,即 HJV、HAMP、TFR2、 SLC40 A1突变是亚洲HH的主要类型[ 1]。
在我国,虽然每年均见血色病个案报道,但发病率不详,遗传背景不明确。个案中进行基因分析的报道屈指可数。在国内血色病患者中,目前尚未见到关于C282Y突变的报道[ 2, 3, 4, 5, 6, 7, 8],仅发现 HJV C321X[ 5]、 HFE H63D[ 6]和 HFE IVS 3+5T>C 3例突变[ 7]。血色病临床表现复杂多样、无特异性,国内50%以上的就诊患者在初诊时被误诊为肝病、糖尿病和扩张型心肌病等[ 9]。
TFR2基因突变导致的3型血色病呈常染色体隐性遗传。2000年,在西西里岛的血色病家系中首先确定该基因突变[ 10]。 TFR2基因位于染色体7q22,由18个外显子组成,编码转铁蛋白受体2蛋白(TFR2)。与TFR1不同的是,TFR2几乎完全表达在肝细胞。TFR2参与肝细胞摄取转铁蛋白结合铁(transferrin-bound iron,TBI),并与HFE结合后,感受血液中铁负载情况,可能通过p38丝裂原活化蛋白激酶(p38 mitogen-activated protein kinase, p38MAPK)/细胞外信号调节激酶(extracellular regulated protein kinase, ERK)和骨形态发生蛋白(bone morphogenetic proteins, BMP)/SMAD(drosophila mothers against decapentaplegic protein)蛋白信号通路调节铁调素(hepcidin)的表达[ 11]。Hepcidin( HAMP编码)是肝脏分泌的小分子肽类激素,是负性铁调节激素。在体内,如果hepcidin升高,血清铁降低;hepcidin降低,铁含量升高。人和小鼠TFR2突变或减低均出现肝脏铁过载并且伴有hepcidin mRNA表达水平降低[ 12, 13]。
3型血色病的临床表现属于“中间型”,即介于青少年型血色病(juvenile HH, HAMP或 HJV突变)和经典型血色病( HFE-HH)之间。本例患者中年起病,发病年龄早于 HFE H63D患者[ 6],迟于 HJV C321X患者[ 5]。临床表现类似 HFE血色病,出现肝功能异常、糖尿病、性腺功能减退、心肌病和皮肤色素沉着。与国内报道的2例 HFE血色病患者(H63D杂合突变[ 6]和IVS 3 + 5T>C 纯合突变[ 7])相比,该患者扩张型心肌病的临床表现更为突出。
根据美国肝病研究协会(American Association for the Study of Liver Diseases, AASLD)血色病诊疗指南[ 14](TS>45%和/或SF升高)进行血色病相关基因检测。患者 TFR2基因中出现2个错义突变(A75V和I238M), SLC40 A1基因中出现V221V同义突变。后者与国内报道[ 15]一致。c.663T>C不改变氨基酸序列,不影响Ferroportin蛋白的功能,所以属于单核苷酸多态性(single nucleotide polymorphism,SNP)。在北京地区汉族人群中,胸腺嘧啶T等位基因频率为20%,胞嘧啶C等位基因频率为80%(rs2304704:http://hapmap.ncbi.nlm.nih.gov/)。 TFR2 A75V和I238M分别位于胞质区和细胞外顶区(Apical)[ 16]。有报道认为这两种突变皆为非病理性突变[ 17](rs41302357/rs34242818)(http://www.ncbi.nlm.nih.gov/gene/7036#variation)。有报道曾在铁超载人群中分别检测出 TFR2 A75V和I238杂合突变[ 18, 19]。本例患者同时出现这两个突变位点,并伴有铁代谢指标异常升高,提示它们是否具有协同作用,影响铁的跨膜转运,导致铁超载,其机制有待于进一步研究。
肝活检以往被认为是诊断HH的金标准,但随着基因诊断的出现,其作用由诊断转向判定肝纤维化(肝硬化)的程度及预后评估。AASLD推荐年龄>40岁、SF>1 000 μg/L或氨基转移酶水平升高的患者应进行肝脏活检[ 14]。本例患者拒绝接受肝活检,因此,无法判断其肝脏铁沉积程度及肝脏受损情况,仅影像学检查提示肝硬化征象。
血色病去铁治疗主要包括静脉放血疗法和药物螯合(去铁胺)疗法。其中静脉放血治疗是目前最有效的方法,它操作简便,费用低廉。若在HH早期进行,可显著降低并发症的发生率和死亡率[ 14]。本例患者有扩张性心脏病并伴有贫血,不适宜行放血疗法,建议去铁胺药物治疗。由于去铁胺费用昂贵,患者放弃治疗,基因检测1个月后死于心肌梗死。
本例患者10年前出现性功能减退和皮肤色素沉着,但未予重视,错过早期诊断和治疗的最佳时期。我国HH发病率低,早期症状无特异性,患者容易被误诊或漏诊为其他疾病,如肝病、糖尿病、扩张型心肌病、内分泌紊乱等疾病,有的患者从出现症状到确诊时间长达30年之久[ 9],因此,提高临床医生对血色病的认识,减少误诊和漏诊,警惕SF和TS同时升高的患者,将有望提高血色病的早期诊断。同时,开展血色病相关基因检测将有助于早日明确我国血色病的遗传基础。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|