{kind=link}
高同型半胱氨酸血症的神经毒性机制研究进展
引用本文
王晓燕, 王伟灵. 高同型半胱氨酸血症的神经毒性机制研究进展. 29(12): 1263-1267
WANG Xiaoyan, WANG Weiling. Research advance on mechanisms of hyperhomocysteinemia neurotoxicity. Labratory Medicine, 29(12): 1263-1267
Permissions
WANG Xiaoyan, WANG Weiling. Research advance on mechanisms of hyperhomocysteinemia neurotoxicity. Labratory Medicine, 29(12): 1263-1267
Copyright©2014, 《检验医学》编辑部
《检验医学》编辑部
高同型半胱氨酸血症的神经毒性机制研究进展
通讯作者:王伟灵,联系电话:021-65415910-6706。
作者简介:王晓燕,女,1977年生,技师,主要从事临床检验工作。
摘要
高同型半胱氨酸血症(HHcy)是由同型半胱氨酸(Hcy)代谢异常引起的。HHcy既是心脑血管疾病的独立危险因素,也与多种神经退行性疾病和精神疾病密切相关。HHcy主要通过血管和神经毒性发挥致病作用。本文就近年来HHcy的致神经毒性作用的机制做一综述。
关键词:
同型半胱氨酸; 高同型半胱氨酸血症; 神经毒性
中图分类号:R446.1
文献标志码:A
文章编号:1673-8640(2014)12-1263-05
Research advance on mechanisms of hyperhomocysteinemia neurotoxicity
Abstract
Hyperhomocysteinemia (HHcy) is caused by abnormal metabolism of homocysteine (Hcy). HHcy is not only an independent risk factor for cardiovascular and cerebrovascular diseases,but also closely correlated with a variety of neurodegenerative and mental diseases.HHcy mainly plays a pathogenic role by vascular and neuron neurotoxicity. In this article, we review the mechanisms of HHcy neurotoxicity in recent years.
Keyword:
Homocysteine; Hyperhomocysteinemia; Neurotoxicity
同型半胱氨酸(homocysteine, Hcy)是蛋氨酸代谢过程中的一个重要中间产物。正常人血浆Hcy水平为5~15 μ mol/L。Hcy代谢异常会引起高Hcy血症(hyperhomocysteinemia, HHcy)。HHcy可分为轻度增高(15~30 μ mol/L)、中度增高(> 30~100 μ mol/L)和重度增高(> 100 μ mol/L)[1]。越来越多的证据显示, HHcy不仅可致动脉硬化, 是心脑血管疾病的独立危险因素之一; 而且还与阿尔茨海默病(alzheimer's diease, AD)、帕金森病(parkinson's diease, PD)、痴呆、认知障碍、抑郁症和精神分裂症等多种神经退行性疾病和精神疾病密切相关[2, 3, 4]。HHcy的致病作用主要包括血管性致病作用和神经毒性致病作用。我们主要对近年来HHcy神经毒性机制的研究进展进行了综述。
一、 Hcy的代谢
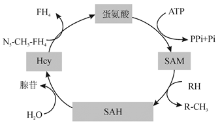
Hcy是蛋氨酸代谢过程中形成的含巯基的氨基酸, 不参与蛋白质的构成, 见图1。蛋氨酸在S-腺苷蛋氨酸合成酶的催化下与ATP结合生成S-腺苷蛋氨酸(S-adenosyl methionine, SAM)。SAM是体内甲基供体, 在甲基转移酶作用下供出甲基后, 变成S-腺苷高半胱氨酸(S-adenosyl homocysteine, SAH), SAH水解生成Hcy和腺苷[5]。Hcy的代谢途径有以下3条[6, 7, 8]。
1.再甲基化途径 叶酸在5, 10-亚甲基四氢叶酸还原酶(5, 10-methylenetetrahydrofolate reductas, MTHFR)的催化下产生5-甲基四氢叶酸, 在蛋氨酸合成酶的作用下, 维生素B12作为辅酶, 5-甲基四氢叶酸作为甲基供体, Hcy获得甲基生成蛋氨酸。这一过程在任何组织中均可发生。Hcy再甲基化的另一途径局限于肝细胞内, 以甜菜碱为甲基供体, 在甜菜碱Hcy转换的酶催化下完成蛋氨酸合成。此过程在肝细胞内完成。
2.转硫化途径 以维生素B6为辅酶, 在胱硫醚β 合成酶(cystathionine-beta-synthase, CBS)的作用下Hcy与丝氨酸结合成胱硫醚。该反应在生理条件下不可逆, 利于Hcy的转运。生成的胱硫醚在γ -硫醚酶作用下裂解为α -酮丁酸和半胱氨酸。后者可再转变为谷胱甘肽或进一步代谢为硫酸盐经尿排出。
3.生成同型半胱氨酸硫代内酯(homocysteine thiolactone, HTL) 在一定生理条件下, Hcy被甲硫氨酰-tRNA合成酶激活, 转化成五元环的硫代内酯。这是Hcy的一种重要衍生物。Hcy与HTL之间可以相互转化。Hcy在酸性条件下易形成HTL; 而在碱性条件下, HTL易发生水解反应形成Hcy。由此可见, 叶酸、维生素B6、维生素B12等摄入不足或蛋氨酸浓度以及代谢过程中关键酶的活性下降都会导致Hcy水平升高。此外, Hcy水平也受遗传因素影响, 如MTHFR、CBS基因突变。目前, 研究较多的主要是轻、中度HHcy, 发现编码MTHFR、CBS的基因发生碱基突变或插入、缺失, 引起相应的酶缺陷或活性下降, 使血浆Hcy水平升高。现已发现MTHFR基因有多种突变类型, 常见的基因突变是C677T。氨基酸替换导致MTHFR基因C677T突变, 使MTHFR活性下降[9]。Huang等[10]研究发现, MTHFR 1298C等位基因也可使MTHFR活性降低和血浆Hcy浓度增加, MTHFR 1298C风险等位基因纯合子与MTHFR 1298A等位基因携带者相比血浆Hcy浓度更高。
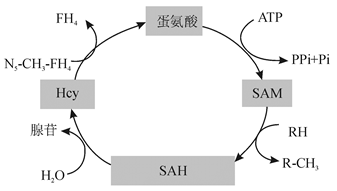 | 图1 蛋氨酸代谢示意图 |
二、HHcy的神经毒性相关机制
1.参与氧化应激反应 HHcy产生神经毒性的一个重要机制是氧化应激。HHcy状态下Hcy可以发生自身氧化, 破坏神经细胞的正常氧化还原状态, 影响其氧化还原信号通路[11]。Hcy的巯基被氧化后产生活性氧(reactive oxygen species, ROS), ROS可攻击神经元细胞膜的多不饱和脂肪酸(polyunsaturated fatty acid, PUFA)和诱导大鼠的脑组织脂质过氧化[12]。ROS对所有生物大分子(如蛋白质、DNA和脂质)来说是高活性的。由于脂肪酸的双键自由基对ROS极其敏感, 因此ROS攻击脂肪酸引发的连锁反应称为“ 过氧化脂质” 反应[13]。在这个过程中会产生很多有害的代谢产物, 如过氧化氢、醛、酮类和醇类代谢物。过氧化物和醛, 特别是4-羟基壬烯醛和丙二醛是参与动脉粥样硬化、炎症扩散、癌症起始和引发神经退行性疾病的关键因素。
2.参与内质网应激(endoplasmic reticulum stress, ERS) 内质网是细胞中蛋白质合成和成熟的主要场所。ERS对决定应激细胞的变化如抵抗、适应、损伤或凋亡有重要作用。HHcy可以通过破坏蛋白质的二硫键诱导ERS, 导致穿越内质网的蛋白错误折叠, 引发神经细胞死亡[14]。PC12细胞株来源于大鼠肾上腺髓质嗜铬细胞瘤, 被广泛用于神经系统疾病的体外研究, 是研究PD的多巴胺神经元的细胞模型。研究发现, 体外PC12细胞经高浓度的Hcy处理后, 细胞活力明显下降, 凋亡率显著增加。ERS时肌醇蛋白1(inositol requiring 1, IRE1)mRNA、肿瘤坏死因子受体相关因子2(tumor necrosis factor receptor associated factor 2, TRAF2)mRNA和蛋白水解酶caspase12 mRNA表达明显上调, 且呈浓度和时间依赖性。显示HHcy可激活ERS通路的IRE1-TRAF2-caspase12转导路径, 导致PC12细胞凋亡[15]。
3.低甲基化途径 SAM和SAH是Hcy的代谢产物。蛋氨酸先生成SAM, SAM去甲基生成SAH, 再进一步脱去腺苷生成Hcy。SAM和SAH浓度是细胞甲基化状态的重要代谢指标。SAM/SAH比值降低通常表示细胞甲基化能力减弱[16]。研究显示, Hcy与SAH浓度呈正比, 与SAM浓度呈反比[17]。SAM和SAH的降低会导致体内许多甲基化酶, 如苯基乙醇胺N-甲基转移酶和儿茶酚-O-甲基转移酶等的活性降低, 从而抑制体内许多重要的甲基化作用。这些重要底物的低甲基化均可导致神经细胞功能受损, 引起认知功能的损害[18]。SAH作为一种强效甲基转移酶抑制剂, 能减慢脑组织中甲基化过程, 使神经元对损害和凋亡更加敏感。HHcy一方面抑制SAH的分解而使其水平增高; 另一方面, HHcy引起甲基供体SAM缺乏, 引起ATP和腺苷水平的不平衡, 导致尿嘧啶错配, 同时影响了嘌呤和胸腺嘧啶修复DNA损伤的能力[19]。
4.细胞外信号调节激酶(extracellular signal-regulated kinase, ERK)途径 HHcy诱导的氧化应激还可以通过ERK途径激活基质金属蛋白酶9(matrixmetalloproteinase 9, MMP-9)。HHcy能增加MMP-9的活性和mRNA表达[20, 21, 22]。大量研究表明, MMP-9在其基因启动子区含有激活蛋白1(activated protein 1, AP-1)位点。此外, AP-1转录活性由ERK-1/2(ERK分为ERK1和ERK2, 统称为ERK1/2)控制。研究表明MMP-9基因表达可能是ERK-AP-1通路的主要激活剂[23]。MMP-9参与细胞外基质降解和血管重塑。然而, MMP-9的异常表达可破坏血-脑屏障, 导致脑微血管渗漏[24], 泄漏的有害分子进入中枢神经系统, 导致细胞损伤和认知能力下降[25]。ERK1/2磷酸化通常与细胞增殖相关联, 并且被视为抗凋亡因子[26]。当叶酸缺乏时, 神经干细胞(neuralstemcell, NSCs)可作为Hcy的重要目标。NSCs是多能细胞, 可分化产生神经元和神经胶质细胞, 其增殖和分化对胎儿大脑发育和成人大脑神经活动来说是至关重要的[27]。叶酸缺乏可导致血浆Hcy浓度增高, ERK1/2活性被抑制, 使NSCs增殖降低、凋亡增加[28]。而Poddar等[29]研究发现, 抑制ERK的磷酸化可以减弱HHcy介导的神经细胞死亡。体外实验表明, 胎儿NSCs暴露于30、100和300 μ mol/L Hcy 4 d后, 磷酸化ERK1/2蛋白表达显著降低, 呈剂量依赖性; 但高浓度的Hcy并没有改变非磷酸化ERK1/2蛋白的表达。由此可见, HHcy可通过抑制ERK1/2的磷酸化来降低胎儿NSCs的增殖。
5.谷氨酸受体途径 Hcy是一种公认的内源性谷氨酸受体激动剂, 与N-甲基-D-天冬氨酸(N-methyl-D-aspartate, NMDA)受体具有高亲和性[30]。NMDA受体在神经系统发育过程中发挥着重要的生理作用, 如可调节神经元的存活, 调节神经元树突、轴突结构发育及参与突触可塑性的形成等。高浓度Hcy可通过活化NMDA受体, 激活钙信号传导途径, 间接增强钙内流, 诱导ROS的产生, 发挥兴奋性神经毒性作用, 导致海马神经元死亡[31]。HHcy时对NMDA受体的激活还依赖于甘氨酸的浓度。当Hcy浓度显著增高(如100 μ mol/L)时, 它才能激活NMDA受体。缺血性卒中、脑外伤和偏头痛时, 脑内甘氨酸浓度明显增高, 此时只需少量Hcy(如10 μ mol/L)即可兴奋NMDA受体, 发挥神经毒性作用[32]。Khedr等[33]对81例脑卒中患者的研究发现, 在其它相关风险因素的存在下, Hcy对认知功能的影响对脑卒中的患者有重大的意义。这就可以解释为什么缺血性卒中时Hcy浓度轻度增高就能够产生明显的神经毒性。
6.蛋白质修饰 随着近年来相关研究工作的不断开展, Hcy对蛋白质的修饰作用方面的成果不断涌现[34, 35, 36]。以蛋白质同型半胱氨酸化为中心的假设理论逐渐受到了世界范围内的广泛关注。蛋白质同型半胱氨酸化是指Hcy及其衍生物由于本身特殊的理化性质对靶蛋白质进行的一种翻译前或翻译后的共价修饰, 使得靶蛋白质的结构和功能发生改变, 进而引起各方面广泛的病理、生理反应[34]。Hcy对蛋白质进行共价修饰的方式是以HTL为代表的N-同型半胱氨酸化。HTL可以以异构肽键与蛋白质的赖氨酸残基结合, 形成N-Hcy-蛋白质, 这一过程称为N-同型半胱氨酸化。蛋白质错误折叠的N-Hcy-蛋白质聚集在内质网内, 通过激活一系列信号通路引起内质网应激[35]。HHcy时Hcy的衍生物HTL生成增加, HTL可以与Tau蛋白的赖氨酸残基结合, 形成N-Hcy-Tau蛋白, 影响Tau蛋白的正常生理功能。Tau蛋白通过与微管蛋白结合形成微管以及维持微管稳定性, 从而促进神经细胞的完整性。N-Hcy-Tau蛋白的形成降低了Tau蛋白对微管蛋白的结合能力, 导致微管蛋白的不稳定, 引起神经元功能损伤[36]。HHcy诱导Tau蛋白的过度磷酸化也是Tau蛋白修改导致神经细胞损伤的机制之一[37, 38]。Hcy可能通过谷氨酸受体途径使蛋白磷酸酶-2A活性下调, 导致磷酸化Tau蛋白水平的增加[38]。AD患者脑中存在大量过度磷酸化的Tau蛋白。过度磷酸化的Tau蛋白每分子可含5~9个磷酸基, 丧失正常生物功能。Tau蛋白的过度磷酸化被认为在AD发病机制中起到重要作用, 蛋白磷酸酶2A能使Tau蛋白双股螺旋形细丝去磷酸, 使Tau蛋白分解、蛋白水解, 并形成各种沉积物沉积在神经元细胞内, 引起神经元损害[39]。
三、结语
综上所述, HHcy通过氧化应激、内质网应激、低甲基化、ERK途径、谷氨酸受体途径、蛋白质修饰等多种途径发挥其神经毒性作用。动物实验也显示了HHcy具有潜在胚胎毒性作用, 影响神经组织发育, 导致神经管缺陷[40, 41]。但HHcy在人体内的影响还有待于进一步验证。HHcy的毒性作用机制相当复杂, 虽然围绕HHcy的神经毒性机制不断有新的理论提出, 但还未形成一个完整成型的理论, 还需要更多的研究证据来补充和完善。HHcy是一个潜在的可逆的危险因素, 及早发现并治疗对于疾病的早期干预具有重要的临床意义。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|