{kind=link}
特定蛋白检测标准化——从CRM 470到ERM-DA 470k及ERM-DA472k
引用本文
唐文佳, 吴炯, 郭玮, 潘柏申. 特定蛋白检测标准化——从CRM 470到ERM-DA 470k及ERM-DA472k. 2012, 27(4): 237-242

Permissions
Copyright©2012, 《检验医学》编辑部
《检验医学》编辑部
特定蛋白检测标准化——从CRM 470到ERM-DA 470k及ERM-DA472k
关键词:
特定蛋白; 标准化; CRM 470; ERM-DA470k; ERM-DA472k
中图分类号:R446.1
文献标志码:A
文章编号:1673-8640(2012)04-0237-06
特定蛋白是指机体内具有某些生理功能, 疾病状态时又起到一定病理作用的蛋白质。特定蛋白结构复杂, 存在多种不同的空间结构和抗原表位。随着认识的逐步深入、检测方法的日渐成熟及各类检测仪器的大量应用, 特定蛋白已成为临床化学重要的检测对象。血清、脑脊液、尿液等不同体液中许多特定蛋白的定量检测已成为临床对疾病诊断、病程监测和疗效评估的重要手段。C反应蛋白(C-reactive protein, CRP)、α 1抗胰蛋白酶(α 1-antitrypsin, AAT)、转铁蛋白(transferrin, TRF)、C3、C4等特定蛋白检测逐渐成为临床化学常规检测, 有的甚至被运用于急诊。为使临床得到更为一致和准确的检测结果, 科研人员不遗余力地进行着特定蛋白检测标准化的各种尝试和努力, 研制出一些较稳定、可靠的参考物质, 在标准化的道路上迈出重大一步。
一、特定蛋白检测标准化及早期发展历史
一般认为, 用于免疫学方法检测的血清蛋白参考物质, 除了需要有良好的均一性、稳定性、可溯源性和结果一致性外, 还需要具备下列条件:(1)参考物质应有足够高的蛋白浓度, 以确保其稀释溶液浓度可以覆盖整个检测范围; (2)基于多数免疫检测使用光学检测法, 参考物质应具有良好的光学通透性, 故参考物质内脂质及脂蛋白含量应尽可能低; (3)使用不同批号参考物质校准后的测量结果须具有一定连续性, 由于临床将根据参考范围和医学决定水平分析检测结果, 因此检测结果应在较长时间内具有可比性[1]。
与检测理化性质明确的小分子物质不同, 相对分子质量大且结构复杂的蛋白或多肽的检测主要通过与参考物质间的比较得出。特定蛋白检测的标准化存在诸多难点, 包括可能存在多种蛋白翻译后修饰, 存在基因变异性, 可能存在对蛋白水解敏感的蛋白配体以及病理状态下体内蛋白水平的不同变化[2]。数十年前开始就有数种不同的血清蛋白一级参考物质面世, 各专业组织和商业机构也研制出不少二级参考物质, 并广泛运用。而这些二级参考物质的定值都是通过不同的方法根据一级参考物质确定的[3]。与此同时, 特定蛋白的检测方法也发生着重大变化, 上世纪70年代开始, 从凝胶免疫电泳法转变为固定时间比浊法, 而后又发展为速率散射比浊法和透射比浊法。检测时间也由最初12 d缩短为数小时甚至数分钟。
1967年, Rowe等[4]研制了数批血清免疫球蛋白制剂, 用于血清蛋白检测标准化。其中批次号为67/86的制剂成为了世界卫生组织(World Health Organization, WHO)首批免疫球蛋白标准品。由国际生物标准品和控制品协会(National Institute for Biological Standards and Control)将该批制剂部分冻干, 并按每瓶中均含有100 U IgG、IgA和IgM对其进行分装, 作为免疫球蛋白的一级参考物质。其余交由英国Wellcome实验室, 研制出后续批次67/95、67/97和67/99[3]。67/95和67/97内蛋白含量通过使用免疫扩散电泳法与67/86比较而得, 67/99内蛋白含量通过计算67/95及67/97单瓶含量均值而得。这3批制剂作为二级参考物质, 由10家专业实验室使用免疫化学法, 以纯化蛋白为参考物质进行定值。由于各实验室间检测变异较大, 使用均值确定定值[2]。
1973年, WHO和国际免疫学协会(International Union of Immunological Societies, IUIS)建议研制新参考物质, 实现性质稳定、无浑浊、可冻干, 且含有数种临床关注的蛋白组分。最后在5种候选制剂中选取一种, 将其部分冻干, 胶塞封存, 作为美国国家参考物质(US national reference preparation, USNRP)。其余部分成为WHO 6种人类血清蛋白参考物质(WHO reference preparation for six human serum proteins, WHO 6HSP)[5]。内含蛋白组分包括前白蛋白(transthyretin/prealbumin, TTY)、AAT、 铜兰蛋白(ceruloplasmin, CER)、α 2巨球蛋白(α 2-macroglobulin, AMG)、TRF以及C3c。此制剂作为这6种蛋白的一级参考物质, 并使用IU作为单位。再次使用免疫扩散电泳法, 以WHO 67/86为该物质进行免疫球蛋白定值, 相应结果成为二级参考物质定值[6]。之后由24家专业实验室为USNRP的各蛋白进行定值[6]。其中免疫球蛋白结果的单位由IU转换为质量, 其中的转换系数与Rowe等在1972年对WHO 67/86的检测结果有所不同。USNRP也因此成为了以质量作为内含蛋白单位的一级参考物质。WHO 67/86高度浑浊, 检测结果非线性, 随着检测技术的进步逐渐不再适用[7]。USNRP逐渐取代了WHO 67/86成为免疫球蛋白一级参考物质[6]。
与此同时, 美国病理学会(College of American Pathologists, CAP)研制出一新参考物质— — 血清蛋白参考制剂-1(reference preparation for serum proteins lot 1, RPSP-1)。这一经过重钙化和脱脂的制剂在其用罄前被许多厂商广泛用作参考物质。随后CAP又研制了RPSP-2、RPSP-3, 并通过与前批制剂相互比较进行定值。然而数年后, 定值结果已与最初根据USNRP得到的设定值产生了偏差。1990年, CAP意识到在新的人类血清蛋白参考物质 (reference preparation for proteins in human serum, RPPHS)研制出前, RPSP-3就将耗尽。为了将定值转换中的变化降到最低, CAP决定使用与制备RPPHS相同的一级参考物质和方法进行定值转换。在此基础上RPSP-4于1991年问世[3]。
随着人们对特定蛋白运用愈加广泛, 质量评估 (quality assessment , QA)体系逐步建立并不断完善, 以评价检测结果的可靠性和可比性。尽管理论上主要的血清蛋白检测在自动化仪器上均已实现标准化, 但来自美国及西欧的QA数据却显示部分蛋白检测结果差异巨大, 差异最大甚至可以达到100%。此外英国外部质量保证计划(UK National External Quality Assurance Scheme, UK NEQAS)的研究显示, 在使用统一参考物质(SPS-01)对检测体系进行校准前后, 实验室间IgG、IgA、IgM、C3、C4及AAT检测结果的变异系数(coefficient of variation, CV)从18.3%(1.6%23.8%)下降至10.7%(7.5%13.6%)。检测结果一致性明显提高(P< 0.001)[2]。故有学者认为实验室间结果的不一致可能是由于所用的不同纯化参考物质缺乏可互通性, 混用不同的二级参考物质或所用参考物质不适用于现行光学检测系统造成的[2]。
据此, 为了提高实验室间检测一致性, 需要一种国际通用的参考物质。1989年国际临床化学学会(IFCC)血浆蛋白委员会(C-PP)开始研制通用参考物质。此制剂中含14种血清蛋白, 包括: AAT、CER、TRF、AMG、TTY、α 1酸性糖蛋白(α 1-acid glycoprotein, AAG)、结合珠蛋白(haptoglobin, HP)、α 1抗胰凝乳蛋白酶(α 1-antichymotrypsin , ACT)、C3、C4、IgA、IgG、IgM及CRP。这一参考物质于1993年通过欧洲共同体标准物质局(European Community Bureau of Reference, BCR)认证, 并给予代号CRM 470。1994年BCR会同CAP将此参考物质全面推广[8]。之后其又被更名为ERM DA-470[2]。
二、CRM 470/ERM DA-470
根据WHO 67/86、WHO 6HSP和USNRP的使用经验, 人们对蛋白参考物质有了更为明确的要求。最重要的是应确保参考物质在检测体系中的性能与血浆标本在常规检测中相似。因此参考物质中的蛋白组分在理学性质上应尽量与新鲜血清相一致, 且在储存过程中保持稳定。通常情况下, 早期的血清蛋白参考物质较新鲜血清更为浑浊, 并且随时间推移其浊度增高。这是由于复钙血浆中纤维蛋白未完全去除造成的沉淀, 这会造成信噪比的下降, 同时降低散射比浊及透射比浊检测的精密度。理想的血清参考物质收集, 需要献血员禁食过夜, 使血液在玻璃器皿内自发凝集, 去除黄疸、脂血和溶血标本, 并且使用二氧化硅微粒吸附残余脂质[3]。
CRM 470是在预处理血清基质中加入纯化蛋白所制成。制备过程中对材料的要求较过去更为严格, 标准包括:(1)选取欧洲区数百名健康个体, 待全血自然凝固后分离血清; (2)所有供血者均需多项病毒抗体的筛查, 同时检测是否存在类风湿因子及单克隆免疫球蛋白, 存在AAT、HP双表型或溶血、黄疸、脂血的标本均将被剔除; (3)使用叠氮化钠作为保存剂, 冷冻运送。使用二氧化硅微粒脱去其他脂质。加入纯化CRP, 将制剂pH值调节至7.2, 无菌过滤后以1 mL/瓶分装, 冻干、封存[3]。
参考物质赋值由欧洲、美国及日本共27家专业实验室共同参与, 定值的操作程序以及统计方法都受到详细规定, 定值采用检测方法包括:免疫透射比浊法、免疫散射比浊法以及凝胶免疫扩散法[3]。
此外, CRM 470较过去的参考物质还有一明显不同, 即不再使用IU作为单位, 而改用mg/L。WHO 6HSP和USNRP虽然出自同一血清池, 但WHO 6HSP以1.3 mL分装(复溶至1 mL); USNRP以1.5 mL分装(复溶至1 mL), 两者复溶后在蛋白浓度上应存在14%的差异。以IU/mL比较两者间含量差异, 平均偏差仅为8.7%, 仅转铁蛋白和免疫球蛋白达到14%左右。改用mg/L则可改善不同标准品复溶后存在体积差造成的使用不便[3]。
作为一个复合参考物质, CRM 470可为临床检测提供便利, 并且使用后显著降低了实验室间的检测差异。1995年对法国众实验室的研究表明, 实验室间和不同检测技术间的批间精密度在使用统一参考物质后有了明显提高(未使用统一参考物质为10.0%24.1%; 使用统一参考物质后为5.8%12.2%), 且准确性也有明显改善[9]。另据欧洲国家质量保证计划(European National Quality Assurance Programs)数据显示, 各实验室大部分蛋白检测结果间差异明显减小, CV的降幅达到5%65%[10]。但同时也有学者认为CRM 470并未实现CER检测的一致性[11]。
至2004年, CRM 470制剂即将用罄, C-PP以及欧洲参考物质和检测协会(Institute for Reference Materials and Measurements, IRMM)着手研制替代物质。为保持各项目检测间的延续性和一致性, 新制剂的制备方式需与CRM 470相一致, 加入蛋白物质后进行赋值[12]。这是一项艰巨的任务, 任何操作过程中的细微变化都可能对制剂的整体性能产生巨大影响。但同时, 新制剂的研制也是对参考物质进行调整和优化的机会。新制剂中将会根据临床需求加入β 2微球蛋白(β 2-microglobulin, β 2-MG), 并去除ACT。
三、ERM-DA470k/IFCC
ERM-DA470具较好的结果一致性, 有效减少不同检测系统间的变异。因此将按照ERM-DA470制备程序制备新的参考物质[1]。
(一)血清池准备
1.筛选献血者 排除糖尿病、黄疸、高血压、心肺及肾脏疾病、妊娠和大强度运动员, 排除高血脂标本, 排除肝炎、人类免疫缺陷病毒(HIV)感染者及出现细菌感染临床症状者。对血清准备有下列要求:(1)餐前采血; (2)将标本收集于干燥容器或聚合物储存瓶中, 不加入任何添加剂或抗凝剂; (3)室温自然凝固, 离心分离血清; (4)-70-80 ℃冻存, -70 ℃运送; (5)进行病毒检测。同时剔除单克隆丙种球蛋白病及类风湿因子> 30 IU/mL的标本。
2.制备血清池 (1)标本于28 ℃融解过夜; (2)血清集中混合; (3)加入叠氮化钠; (4)尽可能除去胆固醇、甘油三酯(TG)、载脂蛋白A(apo A1)、载脂蛋白B(apo B)和总蛋白。
3.转化 将C3转化为C3c。
4.硅胶脱脂 (1)调节pH值至 8.5 ± 0.1; (2)加入NaCl并搅拌; (3)计算硅胶用量(430 mg硅胶/g蛋白); (4)加入硅胶; (5)离心去除硅胶和蛋白沉淀; (6)计算胆固醇、TG、apo A1、apo B浓度, 以确定脱脂是否成功。
5.无菌过滤和渗滤 (1)滤网过滤上清液; (2)渗滤使用等渗NaCl溶液, 以除去三羟甲基氨基甲烷[tris (hydroxymethyl) aminomethane, TRIS]和硅胶产生的游离二氧化硅。
6.防腐和无菌过滤 (1)调节pH值至7.2± 0.1; (2)依次加入叠氮化钠、抑酞酶及苯甲脒盐酸盐水合物; (3)无菌过滤, 同时尽可能除去所有蛋白组分。
7.准备CRP和β 2-MG 使用纯化人类CRP溶液463 mL(占总体蛋白质量的97%, 质量浓度为3.88 g/L)。该物质于-70 ℃冻存, 37 ℃水浴融解同时混匀, 防止出现强烈震荡。β 2-MG的初始材料为冻干纯化重组蛋白。
8.制备最终血清池 (1)溶解所有5个血清池并混合; (2)尽量去除所有β 2-MG及CRP组分; (3)将CRP溶液加入血清池内并搅拌; (4)将重组β 2-MG蛋白溶解于无菌水内; (5)将β 2-MG溶液加入血清池; (6)使用饱和4-羟乙基哌嗪乙磺酸[4-(α -hydroxyethyl)-1-piperazineethane-sulfonic acid, HEPE]溶液调节pH值至7.2± 0.1; (7)无菌过滤; (8)无菌转移3.5 L血清, 并对整体无菌转移物质进行包括无菌测试等的进一步检测; (9)对液体及真空冷冻干燥物质进行检测。
(二)溯源性
血清蛋白参考物质(ERM-DA470、ERM- DA470k/IFCC及 ERM-DA472/IFCC)的制备过程在制剂特性、定值检测等方面严格要求操作的标准化, 确保了在溯源链每一个过程中定值传递的真实性。各参考物质溯源性见图1。
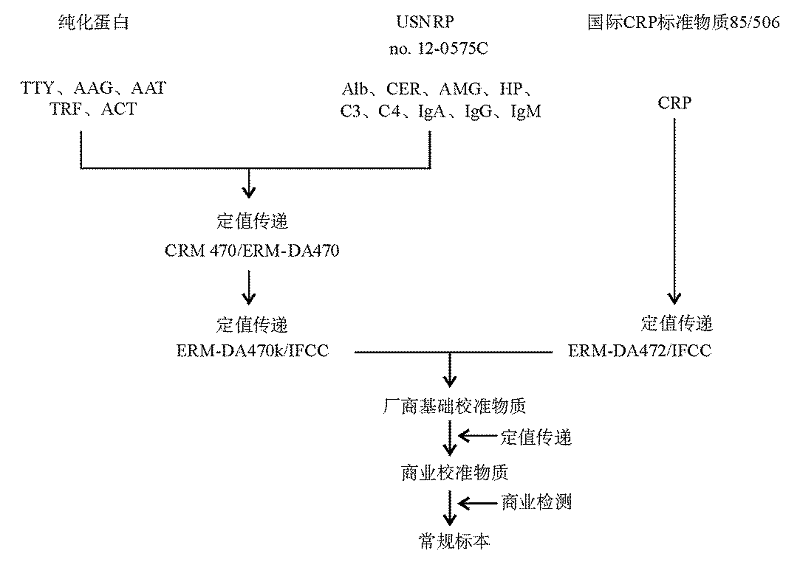 | 图1 参考物质溯源链(注:Alb为白蛋白) |
(三)参考物质定值
人们对参考物质ERM-DA470k/IFCC中AMG、AAG、AAT、Alb、β 2-MG、C3c、C4、CER、HP、IgA、IgG、IgM、TRF以及TTR这 14种蛋白进行了稳定性和均一性评估, 并对除β 2-MG及CER外的12种蛋白进行定值检测, 定值中使用ERM-DA470作为校准品[1]。
实验室检测包括2类检测体系, 即开放定值转换体系和封闭定值转换体系。封闭定值转换系统内使用仪器配套校准品进行校准。将ERM-DA470和ERM-DA470k/IFCC各进行6浓度稀释, 检测2份稀释溶液。对ERM-DA470检测结果与理论稀释浓度进行线性回归分析, 计算斜率比例, 以此得到靶物质浓度。开放定值转换系统中将ERM-DA470稀释溶液作为校准品, 检测ERM-DA470k/IFCC稀释溶液。将检测结果与稀释度进行线性回归分析, 根据斜率计算靶物质浓度[13]。
共18家实验室参与定值, 其中4家使用开放定值转换体系, 12家使用封闭定值转换体系, 另有2家实验室同时使用2种检测系统。所使用检测方法包括免疫散射比浊法(10例)和免疫透射比浊法(8例), 在检测Alb时也有使用可见光光谱测定法[13]。
参加定值的各实验室所得检测数据结果一致性良好, 12种蛋白实验室间CV为1.37%3.02%[12]。CER实验间的平均CV为6.5%, 较其他蛋白更高。这可能是由于铜离子与蛋白结合数量不同造成CER以多种不同形式存在, 其各形式在血清基质中无法长期保持稳定, 以及该蛋白对蛋白水解较为敏感等因素造成。由于不同方法间CER检测结果存在一定差异, 且造成方法学间差异的原因仍不明确。故并未对ERM-DA470k/IFCC进行CER定值。参考物质中β 2-MG的定值将会在今后完成[13]。
ERM-DA470k/IFCC蛋白定值见表1。
| 表1 ERM-DA470k/IFCC蛋白定值 |
(四)ERM-DA470k/IFCC的应用
实验室间和不同检测体系间的变异推动了蛋白检测标准品的诞生。C-PP早期的实验室和方法间检测结果一致性比较的结果极不理想。随着标准品运用的推广, 后期逐渐开展的国际质量保证计划证实了实验室间一致性的改善。据CAP统计数据显示, 各国家间批间检测CV无论是方法内还是方法间都存在一定的改善[2]。截止至2009年, 英、美两国的QA计划数据均显示当前实验室间和检测体系间的CV都有较大幅度下降。此外, 有研究证实了使用ERM-DA470k/IFCC后不同方法学(免疫散射比浊法、免疫透射比浊法)间的检测结果具有良好的互通性[14]。
四、ERM-DA472k/IFCC
CRP由肝细胞合成, 是人体的急性时相反应蛋白。在健康个体中其浓度< 10 mg/L, 但血清CRP水平在应激情况下可升高多达千倍。由于其升高速度快, 响应幅度大, CRP常用于各类炎症、感染及坏死的诊断、预后判断和疗效评估[15, 16]。早期用于CRP检测的方法多为乳胶凝集法[16], 尽管该方法沿用至今, 但其作为半定量方法敏感性不高。免疫透射比浊法、散射比浊法、酶免法和荧光偏振法等更为敏感且真正做到定量检测的检测方法在19世纪70、80年代开始出现[17]。2000年后, 由于学者们逐渐将研究目光转向常规检测限下的浓度水平, 厂商开始研制出更为敏感的检测试剂并且称之为“ 高敏感方法” 或“ 超敏感方法” [18]。随后的研究逐渐发现CRP 不仅可反映全身性的低水平炎症状态, 而且与心血管事件密切相关[19]。
由于在制备ERM-DA470k/IFCC时对原制备实验进行了一定调整, 如加入重组β 2-MG, 改变叠氮化钠浓度和使用渗滤取代透析等, 需要进行预试验以明确各调整可能对试验结果造成的影响[20]。预试验向制剂中加入了重组β 2-MG及纯化CRP。试验数据显示β 2-MG组分在制剂中性质稳定, 并可通用于至少3种常规检测方法, 且加入重组β 2-MG不会对其他蛋白成分的稳定性产生影响。但加入纯化CRP情况有所不同, 冻干和复溶会导致CRP在2种检测方法中出现不完全回收, 使检测值出现约20%的缺失。此外, 新制剂的光学通透性、均一性和稳定性均符合要求[19]。最终新参考物质ERM-DA470k/IFCC中并未加入CRP, 为CRP单独制备了液体冰冻参考物质ERM-DA472/IFCC[2]。
ERM-DA472/IFCC与ERM-DA470k/IFCC使用相同的血清池, 区别在于ERM-DA470k/IFCC最终对血清进行真空冷冻干燥, 而ERM-DA472/IFCC直接进行液态冷冻。
CRP参考物质ERM-DA472/IFCC的定值同样采用与ERM-DA470k/IFCC相同的程序, 8家实验室参与定值, 6家使用开放定值转换系统, 2家使用封闭定值转换系统。各实验室检测CV为4.3%[21]。
ERM-DA472/IFCC的蛋白参考浓度为41.8 mg/L, UCRM为2 g/L。
五、小结
自第1个全球化参考物质ERM-DA470问世以来, 各地血清蛋白检测QA结果的可比性明显提高。新参考物质ERM-DA470k/IFCC在制备时可重现早期的参考物质制作程序, 并可延续参考物质的标准化。数种蛋白的定值结果可确定其定值程序, 可以将纯化蛋白制剂的赋值准确的传递至患者标本, 实现整个检测环节完全可溯源。这一目标是经由IRMM以及IFCC共同努力而实现的, 研究人员希望这一成果可以持续地在各个领域提高蛋白检测的一致性, 为临床提供可靠的数据, 以此提高患者诊疗水平。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|