

{kind=link}
{kind=link}
{kind=link}
临床检验质量规范
临床检验质量规范
作者简介:王治国,男,1964年生,硕士,研究员,主要从事临床检验质量控制方法研究和室间质量评价工作。
摘要
现代质量管理(quality management)涉及的内容要比每天日常工作中执行的简单统计质量控制丰富得多。在质量管理中还包括良好的实验室规范(实践)(quality laboratory practice)、质量保证(quality assurance)、质量改进(quality improvement)和质量计划(quality planning)。这些要素组成了检验医学领域全面质量管理的基本要素[1]。
关键词:
质量规范; 质量管理; 临床检验
中图分类号:R197.323.4
文献标志码:A
文章编号:1673-8640(2012)12-0984-05
Study of result interaccreditation for routine clinical chemistry items in Shanghai
Keyword:
在我们可控制、实践、保证或提高实验室质量之前, 我们必须准确地知道确保满意的临床决策时需要什么样的质量水平。因此, 规定要求的质量即质量规范是建立质量管理所必须的前提条件。见图1。
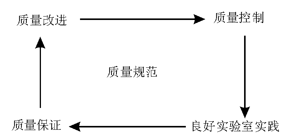 | 图1 质量管理中质量规范的中心作用 |
一、设定质量规范
帮助临床医学决策所要求执行的水平已给出了不同的名称。当前最广泛的名词是质量规范(quality specification)。其他的名词包括质量目标(quality goals)、质量标准(quality standards)、适当的标准(desirable standards)、分析目标(analytical goals)和分析性能目标(analytical performance goals)。
每一方法可由其性能特征进行充分的描述, 其可分为两大类:(1)实用性特征(practicability characteristics)是关于执行程序的详细描述, 包括如要求的技术熟练程度、分析速度、要求的样本量、分析样本的类型等许多方面; (2)可靠性特征(reliability characteristics)是关于方法的科学方面, 如精密度、正确度、检出限和测量范围。
在理想情况下, 实验室程序的每一性能特征都应有质量规范, 特别是可靠性特征中的精密度和正确度(偏倚)。为了执行适当的实验室质量管理体系, 我们必须规定精密度和偏倚以及允许总误差的质量规范。
二、质量规范的使用
通过考虑如何将任何新的分析系统、仪器或方法引入到临床实验室服务, 就能很好地阐述实验室质量管理的许多方面需要客观的质量规范。
1.评价分析系统
在购买检测系统之前及在引入实验室服务之前通常需要进行简单地或详细地评价候选的分析系统或仪器。已有许多优秀的已发表的方案详细地告诉我们如何进行方法评价或确认。 这些数据应该与期望的质量规范进行比较, 以便做出可接受性的判断。
2.建立室内质量控制系统[2]
当引入分析系统或仪器进行服务时, 应建立良好的质量控制系统, 同时引入质量管理的所有其他方面。质量计划是决定检测多少个质控物及什么样的质量控制规则用于接受和拒绝(判断失控)的基础。如果没有详细的质量规范就不能完成此项工作。
3.参加能力验证或室间质量评价计划[3]
对于实验室开展的检验项目有时, 甚至通常是强制性要求参加能力验证或室间质量评价计划。这些计划和方案最好是使用客观设定的质量规范, 使用固定限来判断其可接受性。
三、设定质量规范的层次模式
国际理论和应用化学联合会(IUPAC)、国际临床化学和检验医学联合会(IFCC)和世界卫生组织(WHO)于1999年4月在瑞典斯德哥尔摩举办会议, 讨论在检验医学设定质量规范的全球策略上是否能协商一致。 该份协商一致的声明已发表在斯堪地那维亚临床和实验研究杂志(Scandinavian Journal of Clinical and Laboratory Investigation)[4]的增刊中。“ 声明” 中将可获得的质量规范模式以分级结构方式进行表示。见表1。
| 表1 设定质量规范策略的分级结构 |
质量规范层次模式根据的是《临床化学》杂志早期社论的建议。层次中较高的质量规范模式优于层次中较低的模式。一般建议是适当的模式用于特定的临床目的。当然, 这些建议并不是固定不变的, 因为有可能获得新的和更好的模式。这样就有更好的模式用于特定的专业。
将层次质量规范中提倡的质量规范进行比较的困难之一就是规范的表示有不同的格式。有些规范讲的是精密度, 有些是偏倚, 还有些是允许总误差。
允许总误差质量规范对随机变异和系统变异的联合效果设定可接受准则。许多人建议医生考虑总误差; 质量计划的思想要求使用总误差质量规范; 能力验证和室间质量评价计划使用的固定限也是以允许总误差表示质量规范的形式。因此, 至关重要的是在我们考虑设定质量规范层次及模式结果的实际意义之前如何计算总误差。
四、设定质量规范的策略
在质量规范层次模式中并没有包括所有设定质量规范的策略。 将质量规范设定按其优点进行分层排列, 见表1。然而, 任何特定的策略均存在缺陷。
(一)特定临床情况下的质量规范
理想情况下, 质量规范应由评价分析性能对特定临床决策的影响并以数字方式导出。因此, 对每一试验及每一临床情况, 我们导出的质量规范直接与临床结果相关联。这种方法是处在层次中的最上层。遗憾的是这种方法是非常困难的, 仅能在有限数量的不同的临床情况下对很少的分析物进行计算。
(二)基于试验结果的一般临床使用的质量规范
实验室试验结果可用于许多场合。使用试验结果的2种最主要的临床情况:(1)监测特定患者; (2)使用参考范围进行诊断或发现病例。由此可见一般可应用的质量规范是试验基于生物学变异, 即个体内生物学变异(CVI)和个体间生物学变异(CVG)。层次中的第2层(即表1中的等级2)中的第2种方法是基于医疗观点。通过寻求临床输入, 产生一般的质量规范。然而只有很少的研究是这样做的, 而且一般而言, 做得较差。但这样的观念是很不错的:临床医生使用实验室的试验结果, 这样他们应该能告诉实验室需要什么样的质量。在试验结果常规解释的基础上, 我们计算的质量规范是基于临床医生对一系列短期病例研究作出的反应。因此, 这一策略产生的质量规范是基于感知的医学需求。
(三)从对临床描述的响应中计算精密度
执行数据分析要求的详细计算是很容易的。在此关注的是单个受试者(对象)的结果随时间的变化, 在这种情况下最重要的性能特征是精密度而不是偏倚。
(四)来自专业人员推荐的质量规范
一些国际的和国家级的专业团体已推荐了详细的质量规范。其中有些是关于精密度的, 有些是关于偏倚的, 有些是关于允许总误差的。基于这些建议而广泛采用的质量规范包括:(1)美国国家胆固醇教育计划专家组已发表的关于血脂类分析的精密度、偏倚和允许总误差的推荐; (2)美国糖尿病协会文件规定自身监测血糖系统和糖化血红蛋白分析的质量规范; (3)美国国家临床生物化学科学院已推荐的关于甲状腺激素检测、治疗药物监测以及用于糖尿病和肝功能诊断及监测试验的质量规范(甲状腺激素检测指南正在审核中, 且新的指南建议精密度、偏倚和允许总误差的质量规范最好是基于生物学变异, 如糖尿病和肝功能指南一样); (4)欧洲工作组提议的基于生物学变异用于分析系统精密度和偏倚评价的质量规范; (5)欧洲工作组建议的确认常规方法和用于能力验证或室间质量评价计划材料赋值的参考方法的质量规范(也是基于生物学变异)。
(五)基于法规和室间质量评价的质量规范
一些国家已规定分析性能标准, 认为达到和/或保持认可状态的实验室必须满足该标准。美国临床实验室改进法案修正案(CLIA'88)中规定的允许总误差其实是不精密度加偏倚。当然, 这也只针对一些常见的检测项目。这种策略的优点是CLIA'88很知名, 并易于理解及获得, 甚至在互联网上(www.westgard.com/clia.htm)也可获得。然而, CLIA'88质量要求主要的缺点是基于可达到的标准而不是适当的标准。最近许多关于质量计划的文献均使用CLIA'88质量规范作为允许总误差。世界上许多不同的室间质量评价计划使用不同的技术判断参加实验室的可接受性或可达到的其他性能准则。有些国家分析参加实验室的回报数据, 应用总的或方法组公议值评价偏倚或使用计算的标准差(s)或变异系数(CV)建立可接受的界限, 通常是3s或3CV。但这种情况有明显的缺陷, 因为s或CV仅显示当前方法和技术所能达到的水平。
(六)基于当前技术水平的质量规范
从能力验证和室间质量评价计划组织者那里通常可获得关于分析上实际可达到的数据。如果没有上述的质量规范, 那么可以使用这种通常可达到的当前技术水平作为质量规范。然而, 这样的分析性能不可能真实地反映当前的技术水平。因为分发给参加实验室的样本由于有基质效应而不能真实的像患者样本一样。另外, 实验室工作人员有可能对这些样本采取特殊方式处理, 试图“ 改进” 其性能。能力验证和室间质量评价计划的当前技术水平随时间而变化(并不总是越来越好), 而且取得的性能有可能与实际的医学需要没有关系。
(七)基于生物学变异设定质量规范的策略
在检验医学领域建立的如不精密度、偏倚和允许总误差质量规范的所有策略都有其优点和缺点。当然, 建立质量规范的基本原理应该是:(1)根据医学要求; (2)可用于所有的实验室, 而不考虑实验室的大小、类型或场所; (3)使用简单易于理解的模式产生; (4)受到该领域的专业人员信服并广泛地被接受。从目前情况来看, 基于生物学变异的质量规范可以满足所有这些标准。
五、基于生物学变异的精密度、偏倚和允许总误差的质量规范
(一)基于生物学变异的精密度的质量规范
低的不精密度能减小每一个体试验结果的固有变异性。如果我们知道不精密度是低的, 那么就能够在每分析批中运行较少的室内质量控制样本和使用不太严格的质量控制规则, 并且增加误差检出概率和减低判断结果假失控的概率。这是非常重要的质量计划概念。但重要问题是多低的不精密度才算是好的?增加不精密度将导致增加试验结果变异性。随着分析不精密度(CVA)的增加, 增加变异量上升, 这种上升并不是简单的线性。关于分析变异应< 1/2平均CVI的概念并不是新的, 早在30年前就已提出。如果分析变异< 1/2平均CVI, 则增加到真实试验结果变异性中的变异量大约为10%。仅有10%的分析“ 噪音” 被加入到真实生物“ 信号” 。尽管这是基于经验的判断, 但这种加入分析变异性的量看来是合理的, 这也导致我们要求最好的精密度质量规范是CVA< 1/2 CVI, 即CVI< 0.50。
这种模式在质量规范层次处于较高的位置, 其仅次于评价分析对临床决策的影响。但由于结果分析方法有许多困难, 实际上基于生物学变异分量的质量规范得到许多的支持并被广泛采用已有许多年。因为个体内生物学变异的估计在不同时间和地区是固定的, 所以使用很容易。此外, 关于平均个体内生物学变异数据容易获得也使得计算质量规范变得容易。而且, 在国际和国家指南推荐的许多质量规范(层次的第3层, 即表1中的等级3)也是基于生物学变异[5, 6]。
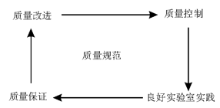
目前, 这种基本概念已扩展:相对于CVI, 增加分析不精密度将增加试验结果的变异性。早期的简单计算已确定:(1)当CVA< 0.75CVI, 则至多有25%的变异性被增加到试验结果的变异性中; (2)当CVA< 0.50 CVI, 则有12%的变异性被加入; (3)当CVA< 0.25CVI, 则最大只有3%的变异性被加入。见图2所示的推荐。
 | 图2 结果变异增加的百分比和分析不精密度与CVI比值之间的关系 |
1.适当的性能(desirable performance)
由CVA< 0.50CVI规定。不同检验项目的生物学变异不同, 其目前所能达到的质量水平也不同。有些检验项目很容易达到上述分析质量指标, 有些则很难。因此, 有必要对“ 适当” 的分析质量指标进行扩展, 在“ 适当的” 上、下分别增加“ 最适宜的” 和“ 最低的” 质量规范。
2.最适宜的性能(optimum performance)
由CVA< 0.25CVI规定。使用这种公式产生的最严格的质量规范应用于由当前技术和方法学容易达到适当性能标准的项目。
3.最低的性能(minimum performance)
由CVA< 0.75CVI规定。使用这种公式产生的不太严格的质量规范应用于当前技术和方法学不易达到适当性能的那些分析项目。
(二)基于生物学变异的偏倚的质量规范
正的偏倚将增加超出上参考限的百分数, 降低超出下参考限的百分数。负偏倚则相反, 但效果相同。从高斯分布的数学上, 我们可以计算当存在偏倚时有多少人将超出每一参考限。
根据医学观点, 对于实验室整个相同的群体范围的基本概念是使用相同的参考范围。这就意味着实验室数据在实验室之间是可移植(转换)的。所以, 患者没有必要每次去不同医院进行重复的实验室试验。如果试验结果仅有很小的偏倚, 那么即使患者看不同科的医生, 使用不同的实验室, 其结果也具有可比性。
但是多大的偏倚可允许这样的参考范围在不同时间和地区之间转换呢?参考范围由CVI和CVG组成。如果分析的精密度是可忽略的, 就可以计算这种“ 组” 生物变异, 如简单的方差相加[(C
对于我们使用的相同组参考值, 则分析偏倚(BA)应< 1/4组的生物学变异, BA< 0.250(C
当BA< 0.375(C
当BA< 0.125(C
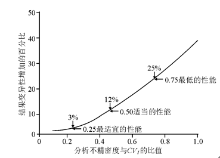
如精密度一样, 这种质量规范应该也有3种水平, 见图3。
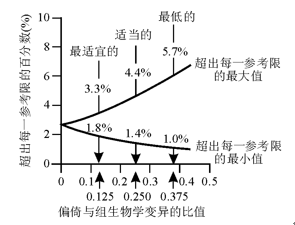 | 图3 超出每一参考限的百分数和偏倚与组生物学变异比值之间的关系 |
1.适当的性能规定为BA< 0.250(C
使用这种公式产生的质量规范通常被认为是适用的, 是最初的、最广为接受的、经常使用的基于生物学变异的质量规范。但是为了满足使用“ 适当” 的质量规范看起来太“ 松” 或太“ 严格” 的那些分析项目, 建议采用以下的质量规范。
2.最适宜的性能规定为BA< 0.125 (C
使用这种公式产生的更为严格的质量规范应用于那些当前技术和方法学容易达到适当的性能标准的分析项目。
3.最低的性能规定为BA< 0.375(C
使用这种公式产生的不太严格的质量规范应用于那些当前技术和方法学不易达到适当的性能标准的分析项目。
(三)基于允许总误差(TEa)的质量规范
最为广泛接受的质量规范是基于生物学变异的层次模式中第2层的质量规范。其他普通的质量规范(适当的性能)分别为CVA< 0.50CVI、BA< 0.250(C
采用3个水平模式是考虑到使用当前方法学和技术不能满足普通的质量规范的那些分析项目。
例如血清中钙和钠的检测。对于这些尚不能达到适当的性能标准的分析项目应采用最低性能: CVA< 0.75CVI、BA< 0.375(C
例如氯的检测, CVI=1.2%、CVG=1.5%, 所以适当的质量规范为CVI< 0.50CVI=0.6%; BA< 0.250(C
也应该考虑当前方法学和技术容易满足普通质量规范的那些项目。例如血清甘油三酯和肌酸激酶检测。对于这些分析, CVA< 0.25CVI; BA< 0.125(C
例如尿素的检测, CVI=12.3%、CVG=18.3%, 所以适当的质量规范是CVA< 0.50CVI=6.2%; BA< 0.250(C
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|