{kind=link}
{kind=link}
{kind=link}
高分辨率熔解曲线及其在分子诊断中的应用
引用本文
沈薇 综述, 傅启华 审校. 高分辨率熔解曲线及其在分子诊断中的应用. 2011, 26(10): 706-709

Permissions
Copyright©2011, 《检验医学》编辑部
《检验医学》编辑部
高分辨率熔解曲线及其在分子诊断中的应用
2. 上海交通大学医学院附属上海儿童医学中心检验科,上海 200127
作者简介:沈薇,女,1977年生,硕士,主管技师,主要从事血液学检验与研究工作。
通过加热使双链DNA解离为单链是DNA基本特性之一,高分辨率熔解曲线(high resolution melting, HRM)正是基于这一特性,采用荧光饱和染料监测熔链过程,从而获得DNA上的相关信息,例如突变、单核苷酸多态性(single nucleotide polymorphism,SNP)等。由于其快速、高通量、低成本,已越来越多应用于临床疾病的分子诊断与分型。
一、HRM技术原理
常规聚合酶链反应(PCR)扩增后,饱和荧光染料可与双链DNA结合,当通过加热升温使双链解离时,荧光染料从局部解链的DNA分子上释放,由于突变、SNP位点双链碱基间不匹配会使双链DNA在此位点首先解开,通过实时监测升温过程中荧光染料强度,从荧光强度与时间曲线上就可以判断是否存在突变或SNP,而且不同位点、杂合子与否、GC含量、扩增子长度等都会影响熔解曲线的峰形。因此,HRM分析能够有效区分不同突变和SNP位点。
HRM采用新型饱和染料如LCGreen,LCGreen Plus,SYTO9,EvaGreen等[ 1],与Rea1-time PCR中被广泛应用的荧光染料SYBR Green I相比有明显的优点。这主要是因为传统荧光染料与DNA结合如要达到饱和,必须高浓度加入,但过高浓度会抑制PCR反应,而饱和染料不抑制PCR反应,因此占据了双链DNA的所有碱基对,此外,当双链DNA局部解链时,游离下来的染料亦不会重新结合到DNA分子上去,所以荧光强度的降低可精准地反映出DNA分子的解链情况。Farrar等[ 2]在测试检出杂合子能力时,认为染料的表现优异依次为LCGreen Plus、SYTO 9、EvaGreen、SYBR Green I。
由于HRM的目的是能对单碱基差异进行区分,所以对温度分辨率的要求相当高。常规熔解曲线升温时每步升高1 ℃,HRM每步升温0.02 ℃~0.1 ℃。HRM是一种高通量的突变筛查和SNP分型的研究方法,同时需要比较多个样品。因此,样品孔之间的温度差异必须小于0.1 ℃,多数常规的定量PCR仪,孔间温度差异多在0.3 ℃~0.5 ℃间,这也是常规定量PCR仪无法胜任HRM分析的主要原因之一。
正是基于以上荧光染料改进与仪器精密度的提高,依靠熔链方法就可实现单核苷酸突变筛查,高分辨率概念应运而生。
二、HRM在分子诊断中的应用
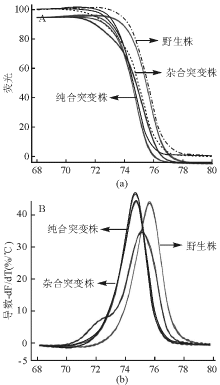
HRM能够在没有标记荧光探针的情况下分析单个碱基的变化。假如扩增子包含有一SNP位点A>C,则可能出现的双链结构为2条纯合双链(A/A和C/C)和2条杂化双链(A/T和C/G),纯合子A/A与C/C熔解曲线相似,但C/C的Tm 值比A/A的Tm值约高0.3 ℃~0.5 ℃ 。而杂合子熔解曲线的图形则不同,其底部宽广即下降温度的变化范围略广。因此,对于单碱基的基因型,杂合型很容易在曲线峰上被检测出来。由于HMR的Tm检测灵敏度在0.1 ℃~1 ℃之间,所以2条纯化双链亦能很容易区分出来,见 图1。
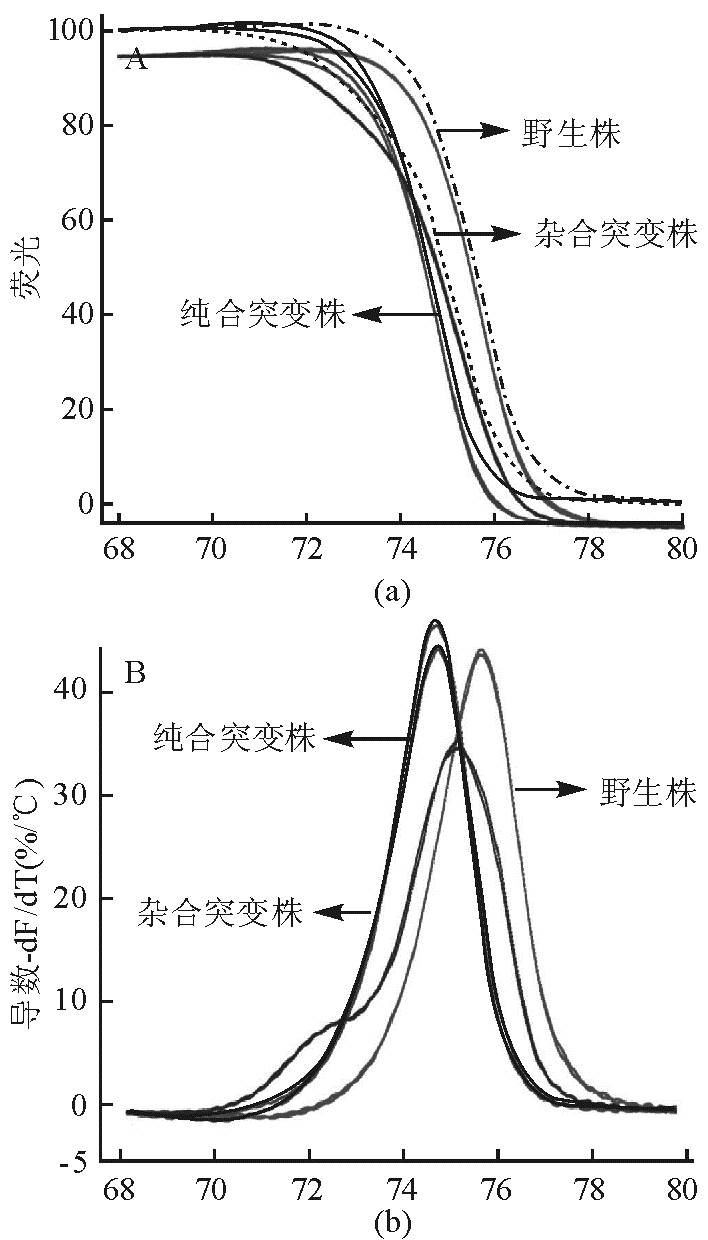 | 图1 原始熔解曲线(a)和衍生图(b) |
1. 突变筛查 Lin等[ 3]运用HRM技术分析了台湾地区122例血友病A家系,结果显示89%(25/28)血友病病例和100%(15/15)携带者都被筛查出,总敏感性达到了93%。Laurie等[ 4]亦用HRM检测了20例存在已知突变的血友病A病例,结果90%(18/20)病例被检测出。
2. SNP分型 SNP分型与连锁分析证实与很多疾病存在相关性,目前亦是研究热点。Kang等[ 5]利用HRM研究了Toll样受体的3个SNP位点,分别是TLR2的597T>C和1350T>C、TLR4的4216G>C,结果显示TLR2的C-C模型可能与过敏性鼻炎有关。Kang等[ 6]亦研究了嗜酸性阳离子蛋白(ECP)基因RNase3的3个SNP位点,分别为-550A>G、371G>C和499G>C,结果显示G-C-G模型与过敏性鼻炎相关( P=0.048),而 G-G-G模型与该病无关( P=0.004)。
3. 微生物的分子鉴定与分型 微生物在自然界种类繁多,变异较快,其相同种属间往往具有相同的表型而造成鉴定困难,而同种微生物分型尤其困难,以往常依赖于血清型鉴定,但特异性较差。HRM的出现,使分子诊断与分型鉴定微生物成为日常工作可能。例如Morick等[ 7]利用HRM调查了以色列地区啮齿类动物的寄生虫状况,并通过rpoB和gtta 2个基因片段和16S-23S基因间隔序列(ITS)将巴尔通体分为8型。Penelope等[ 8]通过对细菌16S rRNA 3个高度多态性区域V1、V3和V6检测,联合生物毒抗原(BT)分析就可对100种细菌进行鉴定。
4. 甲基化研究 HRM同样也可以用于甲基化定量研究,胞嘧啶经重亚硫酸氢盐作用变为尿嘧啶,而甲基化的胞嘧啶则不受影响,因此,甲基化的序列GC含量就高,Tm也就不同,Francesca等[ 9]研究了膀胱癌hTERT和Bcl2启动子甲基化水平,研究发现只要引物设计合适,最少0.025%甲基化DNA就可被检测 。Snell等[ 10]也通过HRM研究了乳腺癌BRCA1基因甲基化,其他还有结直肠MGMT和APC甲基化[ 11]等。
5. 器官移植配型 HRM还可应用于器官移植匹配,此时并不需要准确配型位点,而是将供者与受者样本混合,选择HLA相关外显子,如果匹配,则熔解曲线只有1条,不匹配,则曲线多样化[ 12]。
6. 其他 值得一提的是Seipp等[ 13]对易栓症进行筛查时,用1个PCR反应体系同时对凝血因子V Leiden 1691G>A,凝血酶原20210G>A,亚甲基四氢叶酸还原酶(MTHFR)1298A>C和677C>T 4个位点进行分型,由于这4个序列的Tm值比较接近,因此在设计4对引物时采用富含GC或5'端富含AT引物,使得4个序列长度各异,Tm差别化。Erali等[ 14]也用类似方法对3种曲霉菌进行分型,这种方法时极大节省了时间。
三、非标记探针HRM
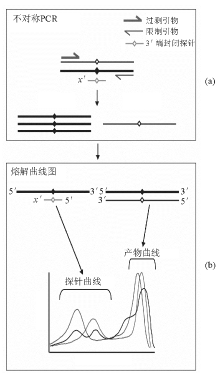
虽然通过HRM可以区分SNP和突变筛查,但是当扩增子序列为1个纯合子突变或存在与疾病无关的多态性位点时,熔解曲线的解读就比较复杂,这时可采用非荧光标记探针方法加以解决。即首先合成SNP位点对应的探针,该探针无需荧光标记,但3'-OH封闭以防止延伸,常用封闭方法为磷酸化。此外在配制PCR反应体系时,2条引物并非等比例,而是按1∶5~1∶10比例加入,1条称为过剩引物(excess primer,EP),另1条称为限制引物(1imiting primer,LP),由于为不对称扩增(asymmetric PCR),因此,需适当增加PCR循环数,一般增加10~20个循环,见 图2。Nguyen-Dumont等[ 15] 研究了ATM基因,详细解释了未标记探针技术。
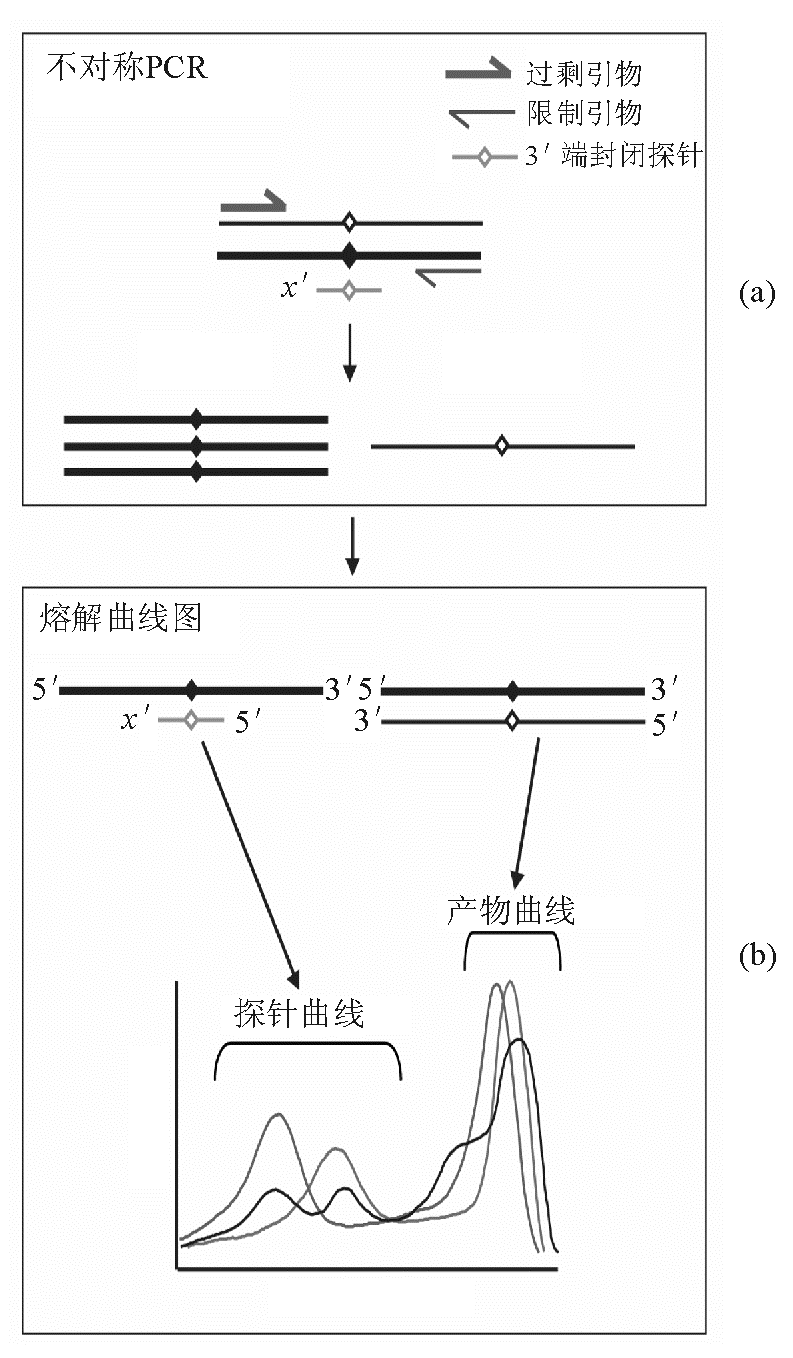 | 图2 非荧光标记探针HRM |
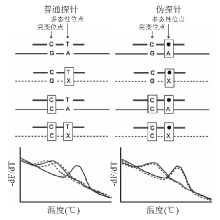
还有2种比较特殊的未标记探针技术,一类为弹回引物(snapback primer),引物末端加1个与靶片段的突变区域互补的尾巴序列,反应体系亦是不对称PCR,带有弹回探针的引物为过剩引物。Zhou等[ 16]针对CFTR第10号外显子设计了2对弹回探针,采用饱和染料LGGreen对100个人的血液进行盲测,检测出第10号外显子的2个区域有7个基因型。另一种特殊的未标记探针,称为伪探针(masking probe),该探针设计成与多态性位点错配,使熔解曲线分析时该位点绝对错配而背景一致,从而有针对分析突变位点[ 17],见 图3。
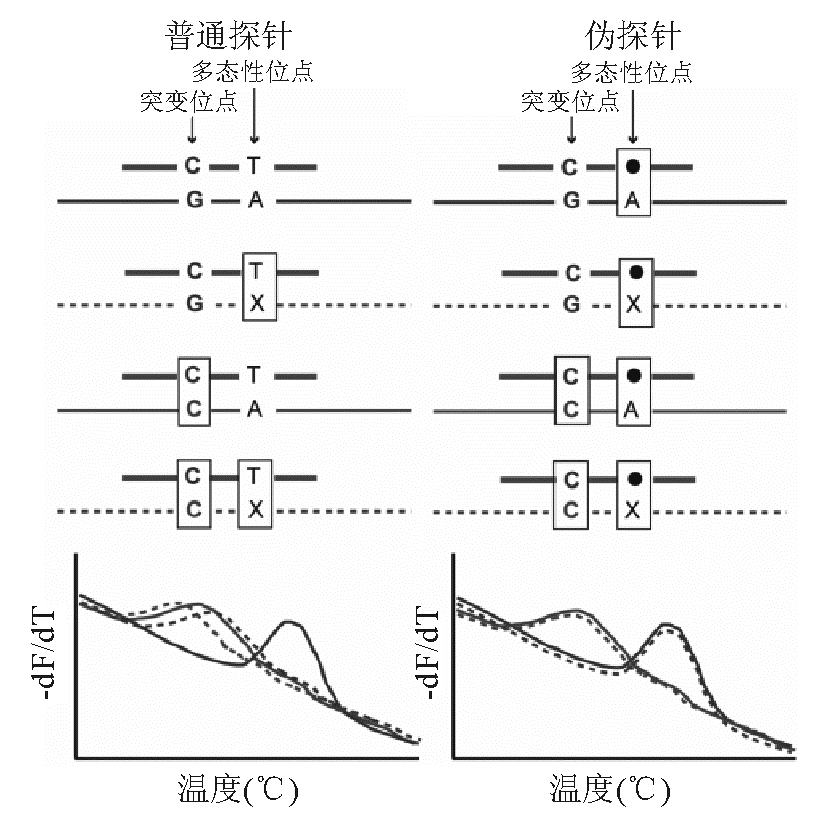 | 图3 伪探针HRM示意图 |
四、HRM的缺点
同所有基于PCR反应基础的技术一样,HRM对于大片断插入或缺失不敏感,此时可采用定量PCR先评估基因含量[ 18]。而从一些对比实验可见HRM技术的敏感性和特异性相对稍差一些,比如与变性梯度凝胶电泳比较[ 19],与荧光标记探针技术(TaqMan)比较[ 20],与变性高效液相色谱分析(DHPLC)比较[ 3]。
HRM对于纯合子缺失和插入也不敏感,此时可通过掺入野生型从而在产物中获得杂化双链,当然也可以设计非标记探针加以解决。HRM对于长片断扩增子错误率明显增高,扩增片段长度最好小于400碱基。
五、总结
HRM是一种相对较新的技术,闭管、简便、快速、经济,但HRM的的分辨率亦需要良好的PCR反应条件、仪器设备和优质荧光染料[ 1],HRM给基因分型、突变扫描和序列匹配带来了简便的检测方法,随着HRM设备和染料的改进,这项技术还会继续迅速发展。HRM是目前最好的突变扫描技术,如果操作得当可以减少95%~99%的测序工量。由于HRM对产物没有破坏性,所以在通过HRM初筛后的PCR产物,还可以再进行测序。这使得其非常适合于测序前的SNP和突变预扫描筛查,相信在分子诊断领域必有新的发展。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|