{kind=link}
{kind=link}
皮下脂膜炎样T细胞淋巴瘤骨髓侵犯伴噬血细胞增多病例分析
引用本文
阎泽君, 刘鸿, 尹新燕, 李秀梅, 范亚敏. 皮下脂膜炎样T细胞淋巴瘤骨髓侵犯伴噬血细胞增多病例分析. 25(8): 669-670

Permissions
Copyright©2011, 《检验医学》编辑部
《检验医学》编辑部
皮下脂膜炎样T细胞淋巴瘤骨髓侵犯伴噬血细胞增多病例分析
作者简介:阎泽君,男,1971年生,学士,副主任技师,主要从事血液学检验工作。
关键词:
皮下脂膜炎样T细胞; 淋巴瘤; 噬血血胞
中图分类号:R446.11
文献标志码:B
文章编号:1673-8640(2010)08-000-00
皮下脂膜炎样T细胞淋巴瘤(subcutaneous panniculitis-like T-cell lymphoma, SPTCL)是指主要累及皮下脂肪组织且与脂膜炎相似的一种原发于皮肤的外周T细胞淋巴瘤, 为皮肤淋巴瘤的少见类型, 累及骨髓者尤为少见。近来我们诊断了1例, 现报告如下。
一、病例
患者, 男, 18岁, 学生, 主因近半年自觉乏力, 周身皮肤结节, 外院查血常规, 发现白细胞及血小板减少, 于2006年3月23日来承德市中心医院会诊。发病以来无发热、纳差, 尿液及粪便正常。查体:一般情况尚可, 无明显消瘦, 全身可触及多个皮下结节, 质硬, 浅表淋巴结未触及肿大, 肝脾触诊未见明显肿大, 双下肢无水肿。血常规:白细胞1.21× 109/L, 红细胞3.77× 1012/L, 血红蛋白107 g/L, 血小板90× 109/L, 中性杆状核粒细胞0.07, 中性分叶核粒细胞0.05, 淋巴细胞0.74, 单核细胞0.09, 不明细胞0.05。
骨髓细胞学检查:增生明显活跃, 粒细胞系统占46.5%, 红细胞系统占19%, 粒∶ 红=2.45∶ 1。粒系比例大致正常, 晚幼粒比例增高, 可见双核, 胞浆空泡。红系比例大致正常, 可见巨幼样变、双核红。片中可见一类不明细胞占0.175, 多散在分布, 少数灶性分布, 该类细胞胞体大, 形状不规则, 胞浆丰富, 灰蓝色不透明, 可见少许粗大紫红色颗粒, 可有空泡, 胞核不规则, 染色质粗网状, 可见核仁1个或数个(见图1)。易见吞噬血细胞现象, 为一类组织细胞, 胞体大, 边缘不整, 胞浆丰富, 淡灰色, 内含吞噬的幼红及幼粒细胞, 或为细胞碎片及空泡, 胞核圆, 常偏于一侧, 染色质粗网状(见图2)。诊断:淋巴瘤骨髓侵犯伴噬血细胞增多。
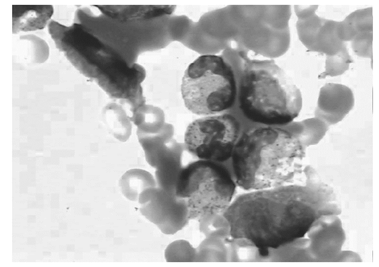 | 图1 骨髓中可见异常淋巴细胞(瑞特染色, 1 000× ) |
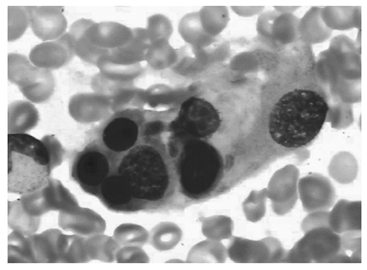 | 图2 骨髓中可见吞噬血细胞现象(瑞特染色, 1 000× ) |
骨髓病理活检:骨髓增生较活跃, 粒红比例减低, 粒系各阶段可见; 红系中晚红为主; 片中可见一类幼稚淋巴细胞散在或灶性分布; 巨核细胞不少。铁染色(+)。考虑淋巴瘤侵犯骨髓。
后建议患者行皮肤结节活检及免疫组化, 结果提示:表皮角化过度, 皮下脂肪层细胞间可见较多幼稚淋巴细胞浸润, 散在及灶性分布。胞体大小不等, 胞浆少, 核不规则, 染色质细, 核仁不明显, 核分裂像易见, 凋亡细胞易见, 较多巨噬细胞, 吞噬红细胞及核碎片(豆袋细胞)。皮肤石蜡切片免疫组化:CD3广泛阳性, CD20阴性, CD56少数阳性, CD68广泛阳性, Gram-B散在较多阳性, 穿孔素散在较多阳性, 细胞毒颗粒相关蛋白-1(TIA-1)广泛阳性。病理诊断:SPTCL。临床诊断:SPTCL骨髓侵犯。
患者诊断后由于经济原因未治疗。
二、讨论
SPTCL为皮肤T细胞淋巴瘤的少见类型, 在世界卫生组织(WHO)新的恶性淋巴瘤分类中定义为一种独立的淋巴瘤类型[1]。本病好发于中青年, Ronald等[2]报道156例患者发病年龄从0.5到84岁, 中位发病年龄38岁, 男女无明显差异, 国内报道以青壮年居多[3]。大多数患者可有体重减轻、发热、乏力、肌痛等全身症状及肝脾、淋巴结肿大。
SPTCL多以皮肤结节为首发症状, 可单发或多发, 本例为多发。皮肤结节主要累及四肢和躯干, 可破损而形成溃疡、出血[4], 而有学者报道[5]溃疡和皮外病变少见。本病可伴发噬血细胞综合征(hemophagocytic syndrome, HPS), 表现为一系、两系或全血细胞减少, 发热、消瘦、肝脾大等症状。
SPTCL的病变原发于皮下脂肪组织很少累及真皮和表皮, 这是SPTCL的一个重要的病理学特点[5]。浸润的细胞可为小、中、大细胞, 但常以某一型为主, 形态类似多形性T 细胞淋巴瘤的瘤细胞, 肿瘤内有多少不等的组织细胞, 可吞噬红细胞和核碎屑( 豆袋细胞) 。本例患者在皮下脂肪层细胞间可见较多幼稚淋巴细胞浸润, 散在及灶性分布。胞体大小不等, 可见豆袋细胞。应用免疫组织化学法对石蜡包埋或冰冻组织标本研究发现, 肿瘤细胞表达T细胞相关抗原如CD3、CD45RO、TCR, 证明为T细胞源性。有学者[6]认为TIA-1阳性对确定细胞毒性淋巴瘤具有重要意义, 因为此类肿瘤可能有更大的临床侵袭性。由于SPTCL常常表达TIA-1, 故TIA-1 抗原的表达对该肿瘤的病理诊断有一定的参考意义。本例患者CD3阳性、TIA-1阳性, 可能与骨髓中出现大量噬血细胞相关。
有关HPS的发生机制国内外报道差别较大, 国内王琳等[6]认为HPS与EB病毒感染相关, 他们报道EB病毒感染率为29.4 %( 5/17), 认为EB病毒相关的SPTCL具有更大的临床侵袭性并常伴HPS, 预后较差 。国外Haque等[7]报道英国的6例患者EB病毒均阴性, 而通过TCR基因分析把SPTCL分为2个亚型, 表达TCR-α β 的3例患者CD8阳性, CD56阴性, 随访16个月后均存活, 而表达TCR-γ δ 的3例患者中2例CD56阳性, 随访16个月后均死亡。认为SPTCL是一种EBV阴性的T细胞克隆性疾病, 来自于表达α β 或γ δ TCR的T细胞, 表达α β 的患者呈惰性病程, 而CD56阳性的病例可能起源于γ δ T细胞, 然后发展成HPS, 预后差。Ronald等[2]报道156例患者, 62%表达TCR-α β , 38%患者表达TCR-γ δ (CD56阳性), 33%(36/109)的患者累及HPS, 中位随访24月后, 48%患者死亡, 中位生存时间27月。而HPS在表达TCR-α β 的患者中占32%, 在表达TCR-γ δ 的患者中占25%, HPS在2个亚型中的发生率无明显差别。然而肿瘤细胞表达TCR-γ δ 和累及HPS者很少存活。EBV的感染率为10%(6/61), 而且6例中有5例来自亚洲, 可能EBV的感染有明显的地域性, 而致使报道不一致。
有关SPTCL的骨髓侵犯, 国内外报道很少, Haque等[7]对6例患者进行了骨髓T细胞基因分析, 1例出现T细胞的单克隆表达。林聪猛等[5]报道8例患者均无骨髓侵犯, 但1例可见骨髓中组织细胞吞噬红细胞现象。本例骨髓中可见异常淋巴细胞, 同时易见噬血现象。
总之, SPTCL具有侵袭性自然病程。预后与TCR基因的不同表达相关, 表达TCR-α β 的患者呈现惰性病程, 而表达TCR-γ δ 的患者CD56阳性, 常伴发HPS, 病情凶险, 预后不良。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|