{kind=link}
反向斑点杂交技术检测HBV基因型方法的评价
引用本文
张韧, 王敏, 符瑞佳, 李娟, 陈建平, 何蕴韶. 反向斑点杂交技术检测HBV基因型方法的评价[J].检验医学, 2015,25(12): 947-951
ZHANG Ren, WANG Min, FU Ruijia, LI Juan, CHEN Jianping, HE Yunshao. Evaluation of reverse dot blot assay for Hepatitis B virus genotyping [J]. Labratory Medicine, 2015,25(12): 947-951
Permissions
ZHANG Ren, WANG Min, FU Ruijia, LI Juan, CHEN Jianping, HE Yunshao. Evaluation of reverse dot blot assay for Hepatitis B virus genotyping [J]. Labratory Medicine, 2015,25(12): 947-951
Copyright©2010, 《检验医学》编辑部
《检验医学》编辑部
反向斑点杂交技术检测HBV基因型方法的评价
作者简介:张韧,男,1977年生,博士,讲师,主要从事病毒性肝炎的诊断和治疗研究。
通讯作者:何蕴韶,联系电话:020-39358007。
摘要
目的 评价用于检测乙型肝炎病毒(HBV)基因型的反向斑点杂交(RDB)方法。方法 应用RDB检测723例HBV阳性标本的HBV基因型。其中对423例HBV序列测序结果清晰可读的样本进一步使用NCBI在线数据库分析、系统进化法分析其HBV基因型,并与RDB法结果进行比较。将样本HBV序列与所用的探针序列进行比对,以评价单个碱基突变对该方法的影响。结果 RDB法检测的检出率为97.6%。RDB检测结果与NCBI在线数据库分析法的一致性为98.8%;与系统进化分析法一致性为100%。与对应探针有1个碱基差异的HBV A型1例、B型12例、C型46例和D型1例均被成功检出。结论 RDB是一种灵敏、准确、有效,且具有很高临床应用价值的HBV基因型检测方法。单碱基的突变不会影响其对HBV的分型结果。
关键词:
乙型肝炎病毒; 基因型; 反向斑点杂交
中图分类号:Q503
文献标志码:A
文章编号:1673-8640(2010)12-0947-05
Evaluation of reverse dot blot assay for Hepatitis B virus genotyping
Abstract
Objective To evaluate the reverse dot blot (RDB) assay for Hepatitis B virus (HBV) genotyping.Methods The HBV genotyping in 723 HBV samples was determined by RDB assay. 423 samples of clear sequencing result were further analyzed by National Center for Biotechnology Information (NCBI) online database, and the HBV genotyping was analyzed systematically. The results were compared with those by RDB. The influence of single-nucleotide mutation on RDB was evaluated, and the HBV sequences were compared with probe sequences.Results The positive detection rate was 97.6%. The concordance of RDB and NCBI online sequencing method was 98.8%, and the concordance of RDB and systematical analysis method was 100%. 1 Genotype A, 12 Genotype B, 46 Genotype C and 1 Genotype D were found 1 nucleotide mismatch to corresponding probe.Conclusions The RDB method is sensitive, accurate and effective for clinical routine HBV genotyping. It is insensitive to single-nucleotide mutation.
Keyword:
Hepatitis B virus; Genotype; Reverse dot blot
乙型肝炎病毒(HBV)根据其基因组大于8%的差异, 可以分为8种基因型。现在越来越多的研究表明, HBV基因型与疾病的发展密切相关, 而且不同基因型的病毒对于抗病毒治疗也有着不同的应答效果[1]。现用于HBV基因分型的方法很多而且各有特点, 然而这些方法都是根据Genbank上已知的序列设计, 现实标本中单个碱基差异就能影响其结果判断, 这也是这些方法不能准确分型的主要原因[2]。应用反向斑点杂交技术(RDB)建立的新型检测HBV基因型的方法, 简便快速, 也被证实能忽略部分标本单碱基突变的影响[3]。然而之前实验中的标本仅仅来源于一个城市的一个医院, 标本量较少且来源局限, 不能准确体现该方法适用的广泛性以及其他地区也普遍存在的、可能是更为复杂的碱基突变对该方法的影响程度。为此, 我们在全国各地医院中收集大量标本, 对该方法在全国适用的可能性进行评价。
材料和方法
一、研究对象
经过荧光定量聚合酶链反应(PCR)(乙型肝炎病毒DNA荧光定量检测试剂盒, 达安基因公司)证实的HBV阳性患者血清723例(HBV浓度1× 103~1× 104拷贝/mL之间的193例, 在1× 104~1× 105拷贝/mL之间的有314例, 1× 105拷贝/mL以上的216例), 由达安基因诊断中心收集, 分别来自于昆明(220例)、广州(70例)、杭州(60例)、乌鲁木齐(55例)、北京(60例)、沈阳(20例)、厦门(42例)、湘潭(27例)和常州(169例)。丙型肝炎病毒(HCV)、EB病毒(EBV)、人类免疫缺陷病毒(HIV)、巨细胞病毒(CMV)核酸阳性血清各2例由达安基因诊断中心收集, 并分别用达安基因公司的相应荧光PCR检测试剂盒证实。
二、实验方法
1.引物和探针 比对Genbank中398条HBV基因组全序列, 选取S基因区进行引物探针设计。反向引物5'端用生物素标记, 探针5'端用氨基标记。正向引物:nt2921-2937, 反向引物:nt3084-3103型特异探针:nt2941-3011; 引物探针序列详见国家专利(申请号:200710026605.8, 公开号:CN101235414)。PCR产物直接测序所用引物为:5'-GAGTATGCCCTGAGCCTGA-3'(nt3084-3102)。
2.DNA提取和荧光定量PCR 每一标本吸取100 μ L血清, 分别加入100 μ L DNA浓缩液混匀, 12 000 r/min(离心半径6 cm)离心10 min, 弃上清。然后加入20 μ L DNA提取液, 振荡数秒充分溶解沉淀。4 000 r/min(离心半径6 cm)离心30 s。100 ℃保温10 min。12 000 r/min(离心半径6 cm)离心5 min, 取上清液-20 ℃冷冻备用。用达安基因股份有限公司的HBV荧光定量PCR检测试剂盒检测各血清标本中HBV的拷贝数, 反应在Applied Biosystems 7000上完成。
3. PCR和RDB 普通PCR扩增在Applied Biosystems 9700上进行。扩增条件如下:93 ℃ 6 min预变性, 然后按93 ℃ 30 s、58 ℃ 40 s、72 ℃ 45 s扩增, 10个循环; 93 ℃ 30 s、56 ℃ 40 s、72 ℃ 45 s扩增, 10个循环; 93 ℃ 30 s、55 ℃ 40 s、72 ℃ 45 s扩增, 15个循环, 最后72℃延伸7 min。PCR产物用纯化试剂盒纯化后送上海英骏生物技术有限公司测序。RDB的杂交过程为:将探针通过氨基结合在硝酸纤维膜上; 将PCR扩增产物95 ℃变性10 min; 变性后的PCR产物与结合在硝酸纤维膜上的DNA探针特异性杂交结合; 洗膜, 洗去未杂交的PCR产物; 已特异性杂交的PCR产物由于其5' 端带生物素基团, 可通过链霉亲和素-过氧化物酶(POD)与之结合, 最后底物四甲基联苯胺(TMB)与POD反应出现可见的蓝色斑点。
4.NCBI在线进行HBV分型 选取PCR测序后返回结果为测序图清晰、无双峰以及其他可疑峰的血清标本423例, 将其测序结果输入NCBI上的在线数据库分析其基因型[4]。
5.系统进化分析 用MEGA 3.0中软件中Neighbor-Joining法对PCR测序结果清晰的423例序列进行系统进化分析。MEGA 3.0系统进化分析能对序列进行聚类, 各类具体属于何种基因型则根据RDB和NCBI在线分型结果予以确定。
结 果
二、RDB检测HBV基因型
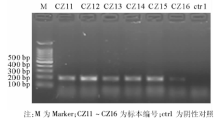
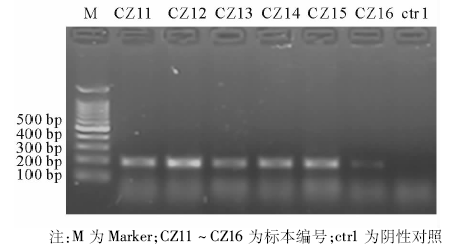
723例标本RDB检测结果见表1。706例成功检测出基因型, 占97.6%(706/723); 未检出基因型的标本17例, 其中HBV浓度在1× 103~1× 104拷贝/mL之间的有14例, 在1× 104~1× 105拷贝/mL之间的有2例, 1× 105拷贝/mL以上的1例。HCV、EBV、HIV、CMV核酸阳性血清均未检出HBV基因型, 且PCR结果均为阴性。
| 表1 HBV阳性标本RDB检测结果表(例) |
三、系统进化分析HBV基因型
选取423例测序结果清晰可读、无双峰的标本, 其序列用MEGA 3.0进行系统进化分析, 结果发现所有423例标本被聚成4类, 分别为HBV A、B、C、D亚型, 其中A型1例, B型161例, C型250例, D型11例; A型和C型基因序列比较接近, B型和D型基因序列比较接近。
四、3种HBV基因型检测方法的比较
423例标本的RDB分型结果、系统进化分析结果、NCBI在线数据库分析结果比较见表2。3种方法结果不同的只有5例, 该5例标本经RDB、系统进化分析都为C型, 而用NCBI在线分型数据库分型为A型。观察测序结果得知其序列完全相同。该序列进一步经过NCBI的BLASTN比对后证实其与C型序列同源性最高(98%)。
| 表2 RDB、系统进化分析、在线数据库分析结果比较表 (例) |
五、探针序列与测序结果的比较
为了验证RDB能否忽略单碱基对分型结果的影响, 将423例标本的测序序列和相应的探针序列进行了比对, 其比对结果见表3。分析结果可知, RDB方法成功检出了所有与探针完全匹配的标本, 同时也成功检出了A型1例、B型12例、C型46例, D型1例与对应探针有1个碱基差异的HBV标本。
| 表3 RDB探针和标本序列比对表 (例) |
讨 论
现在检测HBV基因型的方法有多种[2], 最准确的方法为用PCR扩增HBV基因组全长, 测序之后用系统进化法分析[5, 6]。用于基因分型还可以根据某个基因, 如HBV的被膜基因即S基因[7]进行分型。但测序进行HBV基因分型的主要缺点是不能检测2个或多个基因型的混合感染。如果要克服以上缺点、检出混合感染, PCR之后则需要进行大量克隆的筛选, 如挑选超过100个的克隆才能做到[2]。加上基因测序法费时费力, 因此难以大规模应用于临床。其他进行HBV分型的方法还有聚合酶链反应-限制性片段长度多态性(PCR-RFLP)、型特异引物的单重或多重PCR扩增、线性杂交法、联合单克隆抗体技术法、基因芯片法、荧光定量PCR法等, 这些方法各有特点, 但大多数价格比较昂贵、操作比较麻烦。另外这些方法都是根据Genebank上已知的序列设计, 现实标本中单个碱基差异就能影响他们的判定结果[2]。由于HBV在复制过程中, HBV DNA聚合酶缺乏校正功能, HBV极易发生突变。研究表明, HBV基因组每个位点每年的碱基突变频率[8]为1.4× 10-5~3.2× 10-5; 而在肝移植患者中这一频率更快[9]。因此众多未被报道的碱基差异是这些方法不能正确分型的主要障碍。本研究使用的方法的引物探针来源于大量已知HBV序列的比对; 在方法设计时允许同型序列单碱基的差异, 并且通过杂交温度的摸索实现了忽略单碱基变化这一目的[3]。本研究大规模血清标本的检测结果显示, 该方法成功检出了1例A型、12例B型、46例C型, 1例D型与对应探针有1个碱基差异的HBV标本。说明该方法在实际应用当中应该能有效忽略单碱基突变对分型结果的影响。
经过大样本量的检测, 用本方法检测所有样本的检出率为97.6%(706/723)。而对17例用RDB未能分型的样本分析后, 得知其中14例的HBV在1× 103~1× 104拷贝/mL之间, 2例在1× 104~1× 105拷贝/mL之间, 1例在1× 105拷贝/mL以上。这一结果说明, 当HBV浓度上升时, 其基因型的检出率也相应上升。对未检出样本的分析发现, 1× 103~1× 104拷贝/mL之间的未检出标本中, 8例PCR电泳有产物, 6例无PCR条带; 有PCR条带的8例中, 5例测序结果与探针序列相符合, 3例测序结果不可读; 说明HBV浓度在这个区间的标本有可能造成少量漏检。HBV浓度在1× 104拷贝/mL以上的3例未检出标本, PCR后电泳得知有2例PCR没有条带, 说明可能是因为这2例样本在引物区变异较大, 导致了扩增的失败。另1例PCR条带很亮, 说明扩增成功, 但杂交没有型特异探针显色。经过测序得知, 该例HBV序列探针区域变异较大, 与所有探针都差异2个或2个以上碱基。说明拷贝数在1× 104拷贝/mL以上的标本, HBV序列变异过大并超过了本方法设计的引物探针范围, 可能是不能用本方法成功检出的重要原因。我们还用型特异引物多重PCR法[3]对这些未分型的17例标本进行了分析, 结果只有2例1× 104拷贝/mL以上的标本用多重PCR成功分型为C型, 其余15例2种方法均未能成功分型。这15例中, 在1× 104拷贝/mL以下的13例可能是因为多重PCR法的灵敏度较低, 尤其对于1× 104拷贝/mL以下的标本检出率较低[3]; 而另外2例1× 104拷贝/mL以上的标本可能是因为这2例标本的发生了较大变异, 形成了较独特的病毒序列, 因而导致2种分型方法都不能成功分型。
为了确定所建立方法的准确性, 本研究将本方法的检测结果与基因测序之后的NCBI在线数据库分析、系统进化分析法进行了比较。结果发现, 与系统进化分析法一致性为100%(423/423); 与NCBI在线数据库分析法的一致性为98.8%(418/423)。通过分析5例结果不一致的血清标本发现, 其核酸序列均相同。这5例标本经过系统进化分析、NCBI的Blastn比对, 均证实为C型, 与RDB结果一致; 而测序后的NCBI在线数据库分析却得出是A型的结论。这有可能是因为在线数据库分析法的数据库中, 用于比对的一些标准基因型株涵盖的范围还不够广。中国是HBV高感染率国家, 也是HBV C型突变比较多的国家, 而在所扩增的区域内, HBV A型与C型的序列本来就比较接近, 因此所选择的标准株考虑欠周可能是造成在线数据库分析法部分结果出错的主要原因。
总之, 经过大量样本的验证证实本研究建立的方法应该是一种准确、灵敏、且具有较大临床应用价值的方法。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|