

{kind=link}
{kind=link}
{kind=link}
一例GPⅡb基因突变导致血小板无力症的诊断
引用本文
王静, 李怀远, 郁婷婷, 傅启华. 一例GPⅡb基因突变导致血小板无力症的诊断[J].检验医学, 2015,25(12): 925-928
WANG Jing, LI Huaiyuan, YU Tingting, FU Qihua. A case of laboratory diagnosis of glanzmann's thrombasthenia caused by GPⅡb gene mutation[J]. Labratory Medicine, 2015,25(12): 925-928
Permissions
WANG Jing, LI Huaiyuan, YU Tingting, FU Qihua. A case of laboratory diagnosis of glanzmann's thrombasthenia caused by GPⅡb gene mutation[J]. Labratory Medicine, 2015,25(12): 925-928
Copyright©2010, 《检验医学》编辑部
《检验医学》编辑部
一例GPⅡb基因突变导致血小板无力症的诊断
作者简介:王静,女,1968年生,硕士,副主任技师,主要从事血液学及输血研究。
通讯作者:傅启华,联系电话:021-38625568。
摘要
目的 对1例血小板无力症(GT)患者进行实验诊断,分析引起血小板膜糖蛋白(GP)缺陷的突变基因。方法 通过对先证者凝血常规、血小板计数、血小板聚集功能、出血时间、GPⅡb/Ⅲa(CD41/CD61)含量等项目的检测和血涂片中血小板形态及分布的观察,进行临床表型诊断。通过聚合酶链反应(PCR)扩增结合测序的方法,分析先证者GPⅡb/Ⅲa基因的所有外显子区及其侧翼序列,家系成员仅在相应突变区域进行PCR扩增和测序。同时以100名健康人作为对照进行分析,以排除基因多态性。结果 先证者凝血常规检测和血小板计数正常,但出血时间延长,血小板聚集功能异常;表现为对多种生理性诱聚剂反应低下,但对瑞斯托霉素反应正常,血块退缩不良;流式细胞术检测CD41、CD61含量显著降低;血涂片中血小板散在,无聚集现象。经测序,发现先证者GPⅡb基因23号外显子存在18183A>C杂合变异,导致GPⅡb蛋白Q778P替换。与100名健康对照者测序比较,排除基因多态性,证实其为基因突变。结论 GPⅡb基因的Q778P杂合突变是导致该先证者患GT的原因之一。
关键词:
血小板膜糖蛋白; 整合素αⅡbβ3; 基因突变; 血小板无力症
中图分类号:R446.11
文献标志码:A
文章编号:1673-8640(2010)12-0925-04
A case of laboratory diagnosis of glanzmann's thrombasthenia caused by GPⅡb gene mutation
Abstract
Objective To study the laboratory diagnosis of a patient with glanzmann's thrombasthenia(GT)and analyze the mutation gene causing platelet glycoprotein (GP) dysfunction.Methods The coagulation profiles, platelet count, platelet aggregation test, bleeding time assay, quantity of platelet glycoprotein Ⅱb/Ⅲa(CD41/CD61) and the platelet morphology and distribution observations of the blood smear were used for phenotype diagnosis. All exons of GPⅡb/Ⅲa gene and their flanks were amplified from the proband's genomic DNA by polymerase chain reaction (PCR), and PCR products were directly sequenced to analyze the gene mutation. The PCR products of mutation locus from the proband's family and 100 healthy subjects were directly sequenced to exclude gene polymorphism.Results The coagulation profiles and platelet count were normal, while bleeding time was prolonged and platelet response to various aggregation inducers were impaired excluding restocetin. The clot retraction was deficient. The values of CD41 and CD61 were greatly low by flow cytometer. Platelet was scattered and no aggregative phenomenon in the blood smear. By the DNA sequencing, heterozygous mutation 18183 A>C in exon 23 of GPⅡb gene caused the substitution of Q778P. Compared with the 100 healthy subjects, the possibility of 18183 A>C as a polymorphism was excluded, and it was confirmed as a gene mutation.Conclusions GPⅡb gene Q778P heterozygous mutation is the one of the reasons that cause GT for the proband.
Keyword:
Platelet glycoprotein; Integrin αⅡbβ3; Gene mutation; Glanzmann's thrombasthenia
血小板无力症(glanzmann's thrombasthenia, GT)是一种少见的常染色体隐性遗传性出血病, 其原因是血小板膜上整合素α Ⅱ bβ 3质或/和量的缺陷, 导致患者血小板对多种生理性诱聚剂反应低下或缺如, 影响正常的止血功能。患者终身存在出血倾向, 其出血表现呈现多样性, 即GT的分子缺陷与临床出血症状的严重程度无明显相关性。我们对1例临床诊断为GT的患者进行了相关基因缺陷检测。
材料和方法
一、对象
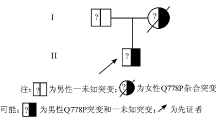
1. 先证者 男, 6岁, 自幼有皮肤瘀斑史, 躯干及下肢内侧经常有自发性出血点和淤青。本次因鼻衄10 d, 伴黑便, 发热39 ℃就诊, 曾输用药物止血敏、冷沉淀物、凝血酶、维生素C、维生素K、血浆等, 均未达到止血效果, 就诊后鼻腔填塞压迫止血。患者父母为非近亲婚配, 母亲因病出血感染而死亡。家系图见图1。家系标本的采集均经受试者或其监护人知情同意。
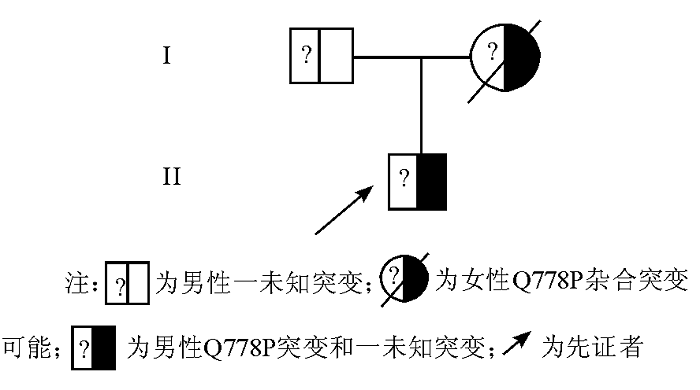 | 图1 本例GT家系图 |
2. 正常对照组 共100名, 男52名, 女48名, 上海儿童医学中心门诊体检健康儿童, 平均年龄(5.7± 2.8)岁。
二、方法
1. 标本采集 采集先证者及家系成员静脉血标本。1份以乙二胺四乙酸二钾(EDTA-K2)抗凝, 用于血小板计数及流式细胞术检测。另1份分3管, 以0.109 mol/L柠檬酸钠1∶ 9抗凝, 第1管用于凝血指标检测, 第2管用于血小板聚集功能检测, 第3管用于抽提DNA, -80 ℃冻存。
2. 凝血相关指标检测 凝血酶原时间(PT)、活化部分凝血活酶时间(APTT)、凝血酶时间(TT)、纤维蛋白原(FIB)的检测采用ACL9000全自动血凝仪, 配套试剂、校准品及标准参比液等为美国IL公司产品。
3. 血小板相关指标检测 血小板计数采用Sysmex 800i血液分析仪, 原装配套试剂、e-check质控品均为日本Sysmex公司产品。血小板聚集功能测定采用Chorono-Log 560 Vs血小板聚集仪, 以2 μ mol/L ADP、0.4 U/L凝血酶、2 μ g/mL胶原、0.5 mmol/L花生四烯酸、0.4 μ g/mL肾上腺素、1.2 μ g/mL瑞斯托霉素为激活剂。血小板膜糖蛋白(platelet glycoprotein, GP) Ⅱ b(CD41)/Ⅲ a(CD61)含量检测采用COULT EPICS XL流式细胞仪, 原装配套试剂、质控品为美国Beckman-Coulter公司产品。
4. 出血时间检测 采用Ivy法, 使用BD末梢采血器和水银血压计。实验中针刺深度为2 mm, 维持加压40 mmHg, 每隔30 s用干净滤纸吸干流出的血液。
5. GPⅡ b/Ⅲ a基因测序 按试剂盒RelaxGene Blood DNA System(北京天根生化科技有限公司产品)说明书提取先证者基因组DNA。聚合酶链反应(PCR)扩增试剂为上海申能博彩生物科技有限公司产品, 包括5 U/μ L Taq DNA聚合酶、10× Buffer、10 mmol/L dNTP。引物参照NCBI GenBank中GPⅡ b/Ⅲ a基因序列(序列号NG008331)设计并由上海申能博彩生物科技有限公司合成, 所用仪器型号为Biometra TGradient。PCR反应总体积50 μ L, 包括1× Buffer、2 U Taq DNA聚合酶以及dNTP(终浓度为200 μ mol/L)、相应引物对(引物终浓度为0.5 μ mol/L)和MgCl2 (终浓度为1.5 mmol/L)。扩增条件:95 ℃ 5 min, 然后95 ℃ 变性30 s, 56~62 ℃退火30 s, 72 ℃延伸30~60 s, 30个循环后72 ℃ 10 min。PCR产物经虾碱酶(上海申能博彩生物科技有限公司产品)纯化后采用ABI 3730 DNA测序仪直接测序。家系成员仅在先证者基因突变区域进行基因检测。
6. 基因多态性排除 对健康对照组DNA进行相应区域的PCR扩增、测序, 进行多态性排除。
结 果
一、凝血常规和血小板相关指标检测结果
先证者凝血常规检测结果:APTT为35.1 s[正常对照组为(35± 10) s], PT为13.2 s[正常对照组为(13± 3) s], TT为15.2 s[正常对照组为(17± 3) s], FIB为4.1 g/L(正常对照组为2.0~4.0 g/L)。出血时间> 40 min, 24 h血块退缩不良。血小板计数为267× 109/L。血小板聚集功能异常, 对多种生理性诱聚剂反应低下, 包括二磷酸腺苷(ADP)、凝血酶、胶原、花生四烯酸、肾上腺素, 其最大聚集率均< 2%, 但对瑞斯托霉素反应正常, 最大聚集率为65%。流式细胞术检测结果显示血小板膜上CD41和CD61的表达显著降低, 阳性率分别为1.36%和5.23%。正常对照组CD41和CD61阳性表达率分别为94.1%± 4.3%和95.0%± 4.6%。血涂片中血小板散在, 大小不均, 无聚集现象, 骨髓涂片巨核细胞成熟障碍。
讨 论
GT由瑞士儿科医生Eduard Glanzmann于1918年首先报道, 为罕见的常染色体隐性遗传性出血性疾病。其病因是GPⅡ b(CD41)/Ⅲ a(CD61)数量减少或质量异常, 导致血小板聚集功能障碍和血块收缩功能不良。
GPⅡ b/Ⅲ a(整合素α Ⅱ bβ 3)通常以复合物形式分布于血小板和巨核细胞表面, 是血小板膜的主要受体, 也是血小板膜上含量最多的膜糖蛋白[1]。在静息状态下, 平均每个血小板约有80 000个α Ⅱ bβ 3分子, 其中70%分布在血小板表面, 其余则储存于膜连接的管道系统和胞质的α 颗粒内。α Ⅱ bβ 3分别由定位于17号染色体的q21-23片段内的ITGA2B和ITGB3基因单独编码。GPⅡ b基因(ITGA2B)包含30个外显子, 编码1 039个氨基酸, GPⅢ a基(ITGB3)包含l5个外显子, 编码778个氨基酸。α Ⅱ bβ 3介导活化的血小板与FIB、vWF因子、纤维连接蛋白、凝血酶、胶原等粘附蛋白结合[2], 导致血小板聚集, 发挥血小板的止血功能。当血小板活化时, 内池的GPⅡ b/Ⅲ a向外释放, 复合物空间构型发生改变, 其上的FIB受体位点暴露与血FIB结合, 发挥止血作用[3]。此外, FIB与GPⅡ b/Ⅲ a的结合还可引起跨膜信息传递, 导致血小板的进一步活化和释放反应, 这些正反馈作用加速了血小板血栓的形成。GPⅡ b/Ⅲ a是钙依赖性异二聚体, 在Ca2+的参与下, α Ⅱ b和β 3亚基以1∶ 1比例构成23 nm的复合物[4, 5], 是一个完整的功能单位。因此其正常生物合成不仅需要各个亚基的正确构像, 而且需要两者相互作用才能形成复合物。当其表达减少或者功能异常时都可能导致血小板不被激活、出血和止血障碍。未形成复合物的α Ⅱ b会迅速降解, 而β 3则可以和α v形成Vitrorectin的受体α vβ 3, α vβ 3可以存在于血小板、巨核细胞、内皮细胞、破骨细胞等表面。
GT可分3型。Ⅰ 型GT血小板表面GPⅡ b/Ⅲ a含量小于正常的5%, 活化的血小板不能结合FIB, 血块缺乏回缩反应; Ⅱ 型GT血小板表面GPⅡ b/Ⅲ a为正常的10%~20%, 活化的血小板可少量结合FIB, 血块回缩异常; Ⅲ 型GT为变异型(结构异常), 血小板表面GPⅡ b/Ⅲ a为正常的50%~100%, 但活化的血小板不能结合或仅少量结合FIB, 血块回缩从缺乏到正常。
本例先证者凝血常规正常, 血小板计数正常, 但出血时间> 40 min, 24 h血块收缩不良。血小板聚集功能异常, 除瑞斯托霉素外, 对多种生理性诱聚剂反应低下。CD41和CD61含量均显著降低, 分别为1.36%和5.23%。血涂片中血小板散在, 大小不均, 无聚集现象, 骨髓涂片中巨核细胞成熟障碍。先证者表型检测符合I型GT诊断。
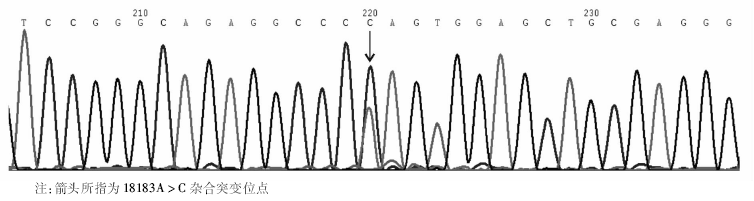
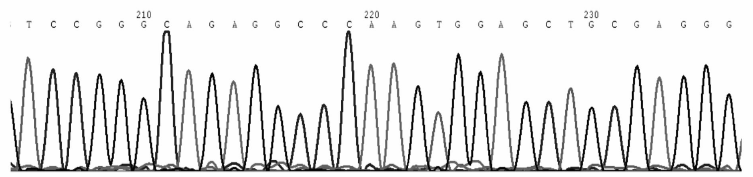
通过对先证者GPⅡ b/Ⅲ a序列的PCR产物直接测序, 发现GPⅡ b基因外显子E23存在18183A> C杂合突变, 导致编码的谷氨酰胺为脯氨酸所替换, 产生Q778P (Gln778Pro)的变异。经检索http://sinaicentral.mssm.edu/intranet/research /glanzmann数据库, 证实该突变已在多个GT家系中有报道[6]。因此基因诊断明确了该突变是导致先证者产生GT的分子发病机制之一。
家系调查发现, 先证者父亲和伯父均无此突变基因, 基于流式细胞术的检测结果和GT的遗传规律, 推断另一突变也存在于先证者的GPⅡ b基因中, 因此推测其因感染出血而病故的母亲可能携带此突变。虽然通过表型检测和GPⅡ b/Ⅲ a基因外显子区测序分析, 可以对血小板膜糖蛋白缺陷症进行实验诊断, 但是由于GPⅡ b/Ⅲ a基因庞大, 结构复杂, 其庞大的内含子区序列的突变也可能对其功能产生影响。在对先证者的GPⅡ b/Ⅲ a基因所有外显子及其侧翼含拼接点的内含子进行测序分析后, 也发现了一些位于内含子区域的基因变异(数据未列)。这些内含子区的基因变异是否会对其转录功能产生影响, 进而影响编码GPⅡ b/Ⅲ a基因的功能, 还有待于进一步研究。从方法学的角度出发, PCR技术结合测序的方法对于一些大片段缺失和突变存在漏检现象。此外, 蛋白翻译后修饰异常、空间结构异常、整合素亚单位的转运异常、整合素的信号传导系统的异常等[7, 8]都能导致GT的发生, 却不一定能找到基因突变。因此, 仅仅通过对外显子区测序分析或cDNA测序分析, 一些遗传性GT的基因缺陷还是不能被完全发现。
GT是一种遗传性出血性疾病, 没有现行有效的治疗指南[9], 目前主要的治疗手段是止血和支持对症处理。血小板输注是最主要和有效的止血方法, 但反复输注血小板可能引起输注耐受, 主要原因是产生了抗血小板GPⅡ b/Ⅲ a的同种抗体, 采用一次性血浆交换去除同种抗体可以取得较好输血效果, 同时局部止血在治疗GT局部出血中具有重要作用。基因治疗GT也正处于积极研究中[10], 希望不久的将来可以通过造血干细胞移植治愈GT。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|