再说说精密度
引用本文
冯仁丰. 再说说精密度. 检验医学, 2014, 29(8): 787-793[FENG Renfeng. Talking about precision again. Labratory Medicine, 2014, 29(8): 787-793] 
Permissions
Copyright©2014, 《检验医学》编辑部
《检验医学》编辑部版权所有
再说说精密度
作者简介:冯仁丰,男,1942年生,主任技师。
摘要
全世界临床实验室对每份患者标本中需要检测的每个分析物都只做一次检测就发出检测报告。这是临床实验室相对于其他分析领域实验室最大的特点,但也是最大的缺陷!如果临床医生和患者对检测报告有怀疑,那么就需要实验室重新做一次检测。重新检测的结果与先前的检测报告一致,这是最理想的结果。为了满足临床和患者的需求,临床实验室定量检验项目的首要分析性能一定是精密度。可惜,国内大多数临床实验室和体外诊断产品的厂商没有对精密度性能予以充分重视。本文呼吁认真学习美国临床实验室标准化协会(CLSI)的EP5文件,并按照文件的实验方案进行完整的精密度实验,以真实、可靠的实验数据建立或验证精密度性能,这才是确保向临床和患者提供有质量的报告的基础。本文也介绍了在验证厂商精密度性能时,比较标准差大小使用卡方检验的含义。
关键词:
精密度; 检测系统; 方差检验; 卡方检验; 实验室内精密度
中图分类号:R446.1
文献标志码:A
文章编号:1673-8640(2014)08-0787-07
Talking about precision again
Abstract
Clinical laboratories around the world reporting determination results deeply depend on only once determining for each analyte for each specimen. This approach only occurs in clinical laboratories in the world, it is the main characteristic of clinical laboratory, however, there also are disadvantages. Relative determinations should be performed again when clinicians and patients have doubts about the reports. The re-determination results are consistent with previous reports, which is the best result. To ensure the quality of determination results and meet the requirements of clinic and patients, clinical laboratories must pay full attention to the precision performance of measurement system, as the primary analytical performance of quantitative analysis. Unfortunately, today's clinical laboratories and in vitro diagnostic manufacturers have not done well precision experiments. This paper calls for the careful study of the Clinical and Laboratory Standards Institute (CLSI) EP5 document, and establishes or verifies the precision performance of each analyte measurement system for determining patient specimen, and this is the key to ensure the quality of determination results for clinic and patients. This paper describes the use of chi-square test for standard deviation when verifying the precision performance from manufacturers as well.
Keyword:
Precision; Measurement system; F-test; Chi-square test; Within-laboratory precision
引言
美国于1988年发布了临床实验室修正法规(CLIA’88)。该法规明确规定了描述一个检测系统分析性能的指标包括7个方面:(1)精密度;(2)正确度;(3)患者结果可报告范围;(4)分析灵敏度;(5)分析特异性;(6)参考区间;(7)其它必须的性能[ 1]。
为什么要将精密度放在检测系统分析性能的第1位?关键原因是临床实验室在检测患者样品时,对样品中的任何分析物或项目都只做一次检测就发出报告!这是全球临床实验室的特殊之处!因此,确保检测结果可靠性的第一前提是检测结果的精密度要好,也就是同一份样品再次检测的结果与第1次是相同的!否则临床和患者均不会对检测结果感到满意。当临床和患者对临床实验室的结果有怀疑时就会要求临床实验室重做一次检测。如果重复检测的结果与上次报告的结果一致,临床与患者均会认为临床实验室报告的结果是“准确”的!两次检测结果一样还不准确吗?其实这并不是准确度好,而是精密度好!由此可见,在任何时候,确保精密度是临床实验室的首要任务。
一、精密度的定义
《2012年国际计量学词汇——基础与通用》[ 2]的概念和相关术语(VIM)中,第2.15节定义测量精密度(measurement precision)[简称精密度(precision)]为在规定条件下对同一或类似被测对象重复测量所得示值或被测量的量值间的一致程度。注意:(1)测量精密度通常用不精密度表示,其值以数字形式表示,如在规定测量条件下的标准差( s)、方差或变异系数( CV);(2) 规定条件可以是测量的重复性条件、测量的综合(intermediate)精密度条件或测量的再现性(reproducibility)条件;(3)测量精密度用于定义测量的重复性、综合的测量精密度或测量再现性;(4)术语“测量精密度”有时用于指“测量准确度”,这是错误的。美国临床实验室标准化协会(Clinical and Laboratory Standards Institute,CLSI)EP5-A2文件中对这些规定条件都做了说明[ 3]。
二、近期困扰我的疑惑
最近,某公司准备将CLSI EP15文件[ 4]写成便于临床实验室使用的应用软件。我认为在形成软件之前必须充分理解CLSI EP15文件的统计含义,绝不能依样画葫芦。否则,当今后有客户遇到不理解的问题进行咨询时,公司只会回答:这是美国CLSI EP15-A文件规定的!但是,该公司的想法促使我再次认真阅读该文件与EP5文件。
CLSI EP15文件的重点是让临床实验室在使用某个新引入的检测系统时必须对厂商声明的精密度和正确度分析性能进行验证。当我再次阅读EP5文件时,发现在EP5文件中已对如何验证厂商声明的精密度提出了实验方案。EP5和EP15文件验证厂商精密度性能的统计做法是一样的。临床实验室使用与某个厂商声明中相似分析物浓度的样品进行精密度实验,得到该分析物项目的不精密度指标( s或 CV)。如该指标低于厂商声明中提供的不精密度指标( s或 CV),则临床实验室的精密度性能验证通过;若该指标高于厂商声明,则临床实验室该分析物项目的精密度性能验证未通过,需要进行统计学检验。经统计学检验后,如果临床实验室的不精密度指标与厂商声明无明显差异,即可认为临床实验室的精密度性能通过验证,反之则为不符合要求。
我反复阅读这两个文件,突然注意到它们在进行精密度( CV或 s)比较时没有使用方差检验(F-test),却使用了卡方( χ2)检验!这令我感到非常突然。我查阅我的统计学书籍[ 5, 6],书中均无答案。这些书籍提供的信息与我了解的一样,从来没有介绍使用卡方检验去判断不精密度( s或 CV)的大小。于是我只能向CLSI专家请教,他们很快就给了回答。经过几次的交流,编写CLSI EP15文件的主席Dr. Neill Carey给出了很好的回答,还怕我不理解,一定要我提出看法。我在EP5文件中看到了相关参考文献,但未找到相关的资料。因此我很希望他能够提供使用卡方检验进行精密度评价的原始文件,但他们回答我说没有参考文献!最后,Dr. Neill Carey给了我一个非常详细的讨论精密度问题的多媒体文件。这给了我很大的启发,让我知道了一个小小的不精密度指标还有许许多多的学问。
我应该是国内最早了解CLSI的人员之一。早在1988年澳大利亚进修期间,看到导师有许多CLSI文件我幸喜若狂。在征得导师的许可后,我复制了几个认为最要紧的标准文件,其中之一即为EP5文件(定量检测方法精密度的评价)[ 7]。回国后,我反复多遍阅读这个文件,而且还看过该文件的每一个修改版本。自认为熟悉和了解EP5文件的我恰恰只着重阅读该文件的前半部分,忽略了后面验证部分,导致不知道以卡方检验比较 s的大小已经有了30余年!
为了弥补我过去的疏忽,近期我重新认真阅读EP5-A2文件。阅读后更感到许多早已经在20年前讲到的问题我却一直没有去理解!尽管EP5-A3及EP15-A2文件将在今年6月发布,但要读到新的文件还需要一段时间。因此,温故知新已不能再等待。
三、临床实验室检测系统精密度性能的重要性
1. 方法学比较中精密度性能的影响 为什么在方法学比较中,在评估检测系统的正确度时EP9文件[ 8]强调对每份比对样品进行双份检测?主要原因是当两个比较方法对每个样品仅做单次检测时会受到方法不精密度很大的影响,使最后正确度性能的评估产生偏移。因此,在进行方法学比较了解系统误差时,如果两个比较方法仅对每个样品只做一次比对,肯定会严重受检测不精密度的影响,导致比对误差过大。
2. 不精密度对线性实验的影响 EP6文件[ 9]是定量检测线性评价的文件。线性评价需对系列已知浓度或稀释关系的样品进行检测。最早的EP6-P文件要求对每份样品进行4次重复检测。只有这样才能了解在实验中每组样品的检测结果是否产生了离群点,也可真实了解是否确实具有线性。这就是精密度性能对线性评价实验的影响。检测范围(measuring range)[可报告范围(repotable range )]定义为测量误差限定在一个特定范围内的分析物测量的一系列值。注意:EP6-P文件的值的范围[分析物(被测量)相应计量单位]在整个范围内符合方法的可接受指标,即非线性、不精密度或其他来源的误差应在确定的限值之内。由此可见,在分析线性的定义中已提及了不精密度对整个检测结果的影响。
3. 不精密度对基质效应评估的影响 EP14文件是评价基质效应的文件[ 10]。为了确认不同类型的样品是否具有基质偏移,EP14文件要求对每个样品至少进行3次重复检测。医学事业的发展促使临床对更多疾病有了深入了解,同时也对临床实验室提出了更高的要求。面临众多分析物、“被测量”和各类疾病的患者,临床实验室必须较以往更重视临床检验基质效应以及不同批号试剂盒间检测结果的差异!
4. 不精密度对分析灵敏度的影响 EP17文件是确定检出限值和定量限值的重要文件[ 11]。对某个检测系统可报告的最低分析物浓度的确定依赖于该检测系统(方法学)的不精密度水平。如果厂商或临床实验室不认真进行精密度实验,将由错误的过小的 s估计出“检出灵敏度”作为自己产品或某个检测系统的最佳检出限值,那么就会对临床诊断造成危害!
四、临床实验室精密度的问题
(一)精密度实验的重复性问题
在所有的分析性能中,精密度性能只要进行重复检测,计算均值( 
1. 分析物浓度 不同分析物浓度会影响不精密度中 s大小,所以应选择适当的分析物浓度进行重复性实验。
2. 基质差异 绝大多数实验室直接使用质控品进行重复性实验,没有意识到质控品与患者天然样品的基质差异。随着临床对实验室的需求不断增加,实验室可以开展的分析物或项目的不断增多,临床实验室需要随时注意在检测某些特殊分析物时真实样品与质控品的显著差异。此时就需使用患者真实样品或其混合物(混合血清)进行分析性能评估及开展质量控制。
3. 精密度估计的重要性 目前,临床实验室无论在评估某检测系统精密度性能还是开展质量控制时均忽略了精密度估计的重要性。更有甚者,直接使用每天质量控制的重复数据计算不精密度。该数据既是该检测系统的精密度,又是开展质量控制的基础资料。但是,我们是否注意到,采用某个检测系统或方法对样品连做20次重复检测,计算 


4. EP5文件[ 3]精密度实验方案的主要做法 EP5文件中提出的“批内不精密度”、“批间不精密度”的实验方案是较客观地反映真实不精密度的估计。可惜,国内大多实验室并没有这样去做。期望大家能花点精力,认真学习EP5文件中的实验方案。EP5文件的做法可以概括成几句话:每天做2批、每批做2份、连做20 d;每批2个结果间的差异就是该样品该批检测不精密度的具体表现。每天上、下午各做一批,最后的数据为20 d、40对、80个。将40对结果间的不精密度差异合在一起就会得到较客观的“批内不精密度”的估计。每批做双份,每天做2批,将每批结果求 









(二)EP5文件能否反映真实不精密度水平
EP5文件这样认真的实验方案是否能很完美地反映临床实验室中检测系统对分析物的真实不精密度水平?30年前,起草EP5文件的专家们就已经想的很多、很远。他们是临床实验室专家、质量管理专家、统计学专家、临床专家和行政管理官员等的完整组合。
1. EP5文件的目的 起草EP5文件的专家们首先考虑的是该文件的目的。EP5文件是为体外诊断(IVD)设备厂商和临床实验室方法的开发者们建立他们方法的精密度能力,也为这些方法的用户们确认性能声明的验证,或度量他们自己的精密度。自动检测程序的用户仅使用最简单的方案验证厂商精密度声明应按照EP15-A2文件(用户展示精密度和准确度性能)进行。该导则对以上情况是完全通用的,因为它包括了考虑精密度估计可靠性的目标。EP5文件也适用于对近期方法做了显著修改的实验室。当使用修改的某个IVD设备或方法时,用户需要确认设备的基本性能特性没有改变,但与原有声明精密度性能进行比较可能不是有效的。典型修改的示例是使用的试剂、样品来源、校准或质控物质或操作程序等不同于厂商使用说明中规定的。
2. 在精密度性能确认和验证上的要求有区别 针对我国大多实验室均为“已经修改了原厂商检测系统组成”的情况,所以不能简单地去验证性能,应该是去确认性能(即建立实验室自行组合的检测系统的性能)或是建立新组合的检测系统的分析性能。因此,应使用EP5文件的方案确认精密度性能。
3. 实验时间长短对精密度性能的影响 EP5文件的实验设计着重考虑了实验时间长短对精密度性能的影响。由于已经考虑到许多因素会影响精密度的结果,所以EP5文件设计方案中已经规定了在一台仪器上,一个批号的试剂、一个批号的校准品、一个操作人员的条件下得到的不精密度水平。在这样的条件下,实验时间成为最重要的因素,用以区分不同的精密度结果。用来叙述与实验时间有关的精密度词语有重复性(repeatability)、批间精密度(between-run precision)、天间精密度(between-day precision)、实验室内精密度(within-laboratory precision)。在这些精密度中,重复性和实验室内精密度一般是最受重视的。EP5文件的实验设计没有合并或特别单独估计其它可能的重要变异来源,如校准品或试剂批号的差异、或技师/操作人员的差异;但是厂商应包括这样的因素以及在不同场地的设备间的变异。影响精密度的其它因素如样品准备、检测材料稳定性、交叉污染和漂移等被包括在本方案中作为实验室内部精密度的来源,但没有分别估计。在EP5文件完整的方案中会使用一个批号的试剂和校准品,但结果的解释(在合适时明确标记)必须包括这个事实,因为这样的结果会低估长期实验室内(或设备内)精密度。引入多批试剂和校准品将增加观察的变异,尽管实验方案中没有允许分别估计这些因素的影响,但会较好地反映检测系统在实际使用中的真实的精密度性能。
4. 我的理解 上述内容明确告诉我们,在估计某个检测系统的精密度性能时,越与日常检验工作接轨就越真实。因此,在估计精密度时不仅需要如EP5文件要求的20 d、40批、80个结果的做法;而且还应考虑多个操作人员轮班操作、换用不同批号的试剂和/或校准品、重新校准等因素,将这些日常的变异因素加入到精密度估计的影响因素中,得到真实的精密度水平。但决不可加入随意更换试剂厂商、随意调整计算因子等不规范行为造成的影响因素!
五、验证实验室精密度性能
1. 验证精密度的问题 当前的实验室管理要求中规定实验室在使用检测系统检测患者样品、发出检测报告前必须验证检测系统的分析性能,确认其分析性能符合厂商说明书中的声明(claim)。因此,检测系统厂商建立的分析性能必须要比临床实验室的常规检测更加严格。CLSI EP5文件中对厂商建立分析性能也提出了相应的建议。国内外有那么多试剂、仪器组成的各种不同的检测系统均被我国CFDA批准准许销售。但是各家厂商在建立分析性能的严密性上各不相同。按照美国政府的认识,无论FDA对IVD产品的管理还是CLIA对临床实验室的管理均代表政府行为。因此,临床实验室使用的IVD产品必须得到FDA的批准;临床实验室在使用前必须按照CLIA要求对分析性能进行验证(验收)。验证的指标就是厂商的分析性能声明!所以,厂商的分析性能指标就成为国家要求实验室“遵守”的要求!那么在验证中,临床实验室得到的精密度性能如何与厂商声明的精密度性能进行比较呢?现在的做法很简单:在与厂商精密度指标采用的分析物浓度几近一致的条件下,只要临床实验室得到的 s低于厂商的指标,该分析物项目的精密度性能即被验证!但实验室管理部门从来没有考虑过:厂商说明书中的精密度指标是否符合要求?实验室的实验方案是否符合要求?厂商在建立分析性能时大多会选择简便的方法。而且为了说明产品质量的优秀,相关数据也会选择偏小的进行报告。因此,临床实验室越是认真的进行相关实验,得到的精密度数据就越客观,但精密度性能通过验证的机率也越小!所以没有临床实验室愿意严格按照CLSI EP5文件的要求进行相关实验。这种做法是当今临床实验室的严重弊病!也正因为如此,当临床实验室的精密度指标与厂商的精密度指标进行比较时,两个精密度均不确切,很难说明究竟哪个精密度更真实!
2. 专家的认识 编制CLSI EP5文件的专家很早就认识到:仅仅将2个 s放在一起比较数值是不可靠的。值得注意的是,这些比较是一个浓度下的点的比较!在厂商没有说明得到 s的实际实验方案,但产品又获得政府批准的情况下,只能假设厂商 s的自由度为无穷大!临床实验室的验证实验也做的很简单,最终导致得到的精密度只能是“真实 s”的一个很粗糙的抽样,不可靠的因素很多。因此,在临床实验室的精密度指标与厂商比较时,必须调整检测的自由度(degree of freedom),使两者标准差的比较在相应估计的自由度下进行。
3. 我的肤浅认识 Westgard 等[ 12]提到,在方法评价研究中, F检验有时候用于比较检测方法和比较方法的方差。方差简单地说就是 s的平方。简言之, t检验用于系统误差或准确度; F检验用于随机误差或精密度。为了进行 F检验,取实验方法和比较方法 s的平方值,将较大的方差作为分子,较小的方差作为分母,求 F值。公式为 F= 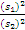
4. CLSI专家的启示 在比较临床实验室按CLSI EP5文件和EP15文件得出的精密度结果与厂商声明的一致性上,CLSI导则使用了卡方检验,而不是使用 F检验,这是基于以下的考虑:在厂商说明书的精密度表中,“声明”代表了有关行政机构批准的标记(labeled) s。我对这段话的理解是厂商的 s经FDA批准后已经成为实验室必须要实现的目标。但是,厂商没有明确说明他们的精密度具体是怎样得到的,因此不可能按 F检验的要求确定与厂商估计有关的自由度。同时厂商也没有明确说明在重复性实验中是否进行多个试剂批号、多个仪器的实验。所以临床实验室在处理厂商说明书中的值时没有方法可供选择,但又处于必须去评估估计自由度的境地。再者,正如前所述,临床实验室实验得出的 s也有很多不足的地方。那么,如何来比较临床实验室和厂商声明中的精密度?一个做法是调整比较时的自由度。一般来说,自由度越小(即重复检测次数越少),得到的抽样 s值的可信性就越差,即 s的可信区间很大。 F检验的自由度由两个进行比较的实验各自的重复次数决定,无法进行调整。所以在统计功效上的能力较差。相比之下,卡方检验在比较 s的差异上具有较好的统计功效。这也是我以往不懂的地方,至今对此概念依然非常模糊。请教工业上的有关专家,他们告知在统计学上确实有使用卡方检验比较 s的方法[ 13]。
5. 与厂商声明的比较 临床实验室通过上述实验得到的精密度估计应与厂商声明的精密度进行比较。可以采用以下叙述的卡方检验进行统计。为使用这个方法,厂商的性能声明中被表示为一个点的估计(即一个 s)。重复性和综合精密度的估计应分别进行比较。性能声明标准差( σr)须注明。卡方检验使用了临床实验室和厂商重复性估计的平方,必须知道临床实验室估计的批内方差( 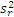
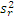
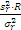
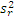
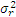
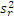
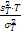
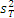
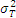
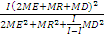
六、总结
1. 精密度性能是医学检验的基础 由于医学检验的特殊性,全球临床实验室在对每份患者标本进行分析物检测时,如无特殊情况,均对每个分析物只做单次检测就发出检验报告。临床和患者对检测结果是否满意取决于实验室再做一次检测的结果与前次结果是否一致。因此重复性是临床实验室检测最重要的方面,千万不可忽视。
2. 认真学习EP5文件 30余年前,美国临床实验室标准化委员会(NCCLS)已经考虑到多方需求和影响精密度的各个因素,编写了EP5文件。完成一个重复性实验的方式有多种。为了患者样品检测结果的可靠性,必须充分考虑众多影响因素,以适当的方式得到可靠的精密度估计。我已在临床实验室领域内工作和学习50年了,但是还有许多方面需要我继续努力学习的。我将精密度视为临床实验室的关键所在。几乎所有的临床检验问题均由此产生。因此关注和重视精密度性能是解决临床检验问题的基础和起点。我愿意与临床实验室的同道们一起,为提高临床实验室的地位而努力。临床实验室的地位只能靠自己去争取,依靠自身的提升,得到临床和患者的认可。在与CLSI专家的交流中,Dr. Neill Carey告诉我:“EP15-A2文件中的方法比较实验将在EP15-A3中删除;在EP15-A3文件中,我们不再比较两个方法的精密度估计。我们做了实验,评估EP15(A2和A3)文件中一个方法的精密度。我们比较了观察(计算)得到的 s与厂商声明的 s。为了了解观察到的 s是否真的超过厂商声明的 s,我们计算了厂商声明中 s的确认限值,将观察到的 s与确认限值做比较。如果观察到的 s超出确认限值,即观察到的 s确实高于厂商声明的 s,仅有5%的可能性是因为随机所致。”这两个文件将于今年6月正式发布。由于上述的实验结果,专家们在新的文件中不再要求临床实验室对验证中实验精密度超过厂商声明进行统计检验。这无疑是简化了验证程序。
附录:EP5文件实验室内精密度估计
实验方案:对稳定的实验样品每天做2批实验,批间相隔的时间不少于2 h;每批实验均对样品做双份检测;共做20 d。一共有40对、80个结果。每对结果间的差是每批的批内差。20 d内共做了40批,这些差值的归纳客观反映了较长时间周期内的批内不精密度。对每批实验的双份结果以 






下述统计公式基于上述双份检测得出的 s。
1 . 批内不精密度的标准差( s批内)
s批内 = 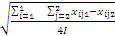
式中:I为实验总天数;i =1为总和从第1天开始计;2表示1 d内实验的批数,j表示批数的序号(只能是1和2,因为每天做2批;j=1表示总和要从每天的第1批开始计);4I表示组成总和的实验数据的个数(如果做了20 d,则 I为20、4 I为80)。
2 . 批间变异的估计值(A)
A= 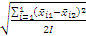
式中:I为实验总天数;i=1为总和从第1天开始计; 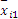
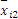

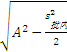
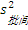
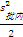
3. 天间变异估计值(B)
B= 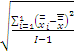
式中:I为实验总天数;i=1为总和从第1天开始计; 

4. 天间标准差(s天间) 由B值可求s天间,公式为s天间= 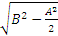
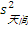
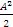
5. 实验室内标准差(s总) 最后,可以按照方差和的方式将批内s、批间s和天间s结合起来求出s总。公式为 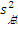
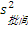
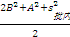
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|